Klinische Genetik Dr Gyrgy Fekete II Universitts Kinderklinik













 n Informative morphogenetische Varianten Normale Organ-Funktionen, ästhetische Veränderungen ohne pathologische Wirkung")

 n Klinodaktylie")
")


")
 Ohren Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika")
")
 n Verzögerung")
 England, Earlswood Asylum n Down- Syndrom")


 ( Atrio- Ventrikular- Kanal, Fallot -")
 Präsenile Demenz, Alzheimer • Mentalretardierung Kleinwuchs Häufige Infekte")
: akrozentrische Chromosomen,")

 n")


- Syndrom John Bruce Beckwith (1933 - ), Pä P")



 Klaus Pätau (1908 – 1975), Humangenetiker, Deutschland, USA, 1:")
 Hexadaktylie")

 John Hilton Edwards (1928 -2007) England, Humangenetiker Semmelweis Egyetem, II.")

 Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika")
 Deutscher Kinderarzt, Henry Hubert Turner")
: Lymphödem / Hände, Füsse, Pterygium colli Semmelweis Egyetem,")
, USA, Endokrinologe n Keimdrüsenunterfunktion im Pubertäts.")
 n Einige Mikrodeletionen schliessen dominante und geschlechtsgebundene")

, Deutscher Humangenetiker, Kurt Hirschhorn")



, Schweiz, Kinderarzt u. Endokrinologe,")



, England, Kinderarzt Semmelweis Egyetem, II. Sz. Gyermekgyógyászati")




 Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika")

 genomische Hybridisierung (CGH) Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika")




 Kyphoskoliose Senkfuss Hüftgelenksarthrose")
 (lange Finger +")


, 15 q 21. 1 Über 200")




 Idiopathische Pankreatitis")
 - Gen Mehr als 2000 Mutationen ,")

 Fehlen des Dystrophins")

")
 Milder als DMD, späteres Beginn Kein Fehlen , sondern Änderung der")
 Rett – Syndrom")
, Xq")
 Geistige Retardierung Epileptische Anfälle Neurologische")
- Slides: 91
Klinische Genetik Dr. György Fekete II. Universitäts - Kinderklinik Semmelweis Universität Budapest, Ungarn
Die klinische Bedeutung und wichtige Entwicklungen n n n Genetische Erkrankungen : ~ 11% der Geburte Pränatale Diagnostik Vorgeburtliche Untersuchungsmethoden: Ultrasonographie, Karyotyp – , biochemische- und molekulargenetische Analysen, Chorionzotten – Biopsie (CVS), Amniozentese, mütterliche biochemische Marker Molekulargenetik Genetische Beratung – Die Rolle des Arztes Prädiktive und präsymptomatische Diagnostik Erkennung von teratogenen Noxen
Das Ziel der angewandten klinischen Genetik Hilfe zur individuellen Entscheidungen n Neue, spezifische Methoden n Nicht direktive genetische Beratung n
Der Hausarzt soll an genetische Ursachen bei einer Krankheit denken Die klinischen Symptome sprechen dafür n Die gleiche/ ähnliche Krankheit bei Angehörigen n Die Manifestation ist früher als üblich n Eine bestimmte Bevölkerungsgruppe n Eine sehr seltene Krankheit n Verwandtenehe n
Genetische Erkrankungen/ medizinische Disziplinen Kinderheilkunde n Gynäkologie - Geburtshilfe n Innere Medizin n Gerichtsmedizin n Kardiologie n Neurologie, Psychiatrie n Immunologie n
Onkologie, Hämatologie n Infektologie n Endokrinologie n Augenheilkunde n Dermatologie n Rheumatologie n Orthopädie n Chirurgie n
Warum sollen Ärzte über die klinische Genetik informiert sein? n n n Die meisten häufige Krankheiten haben genetische Assoziationen Genaue diagnostische Methoden Nosologische Klassifizierung – genetische Grundlagen Genetische Polymorphismen – spezifische Krankheitsdispositionen, Risiken Individuelle Therapie
Genetische Fragestellungen Anzahl der Patienten und Untersuchungen/ Jahr/ II. Uni. - Kinderklinik Budapest n Genetische Beratung: ~ 430 Familien n Zytogenetische Untersuchungen: ~ 550 n Molekulargenetische Analysen : ~ 520 n Zytogenetisches Labor und Genetische Beratung seit 1961
Die Stelle der klinischen Zytogenetik Genetik n Humangenetik n Medizinische Genetik n Klinische Genetik: die genetischen Kenntnisse des behandelden Arztes (Diagnose, Behandlung, Prävention). Eine der Methoden ist die klinische Zytogenetik, die Wissenschaft der Chromosomenaberrationen n
Historische Daten 1882: Flemming - Chromosom = farbiger Körper 1903: Sutton, Boveri - die Chromosomen tragen die Gene 1955: Joe- Hin Tjio und Albert Levan- 23 Paare humane Chromosomen 1959: Jerome Lejeune- ein überzähliges Chromosom ist die Ursache des Down Syndroms 1969: Turbjörn Caspersson, Lore Zech – Quinacrin - Bandenfärbung
Bedeutung der angeborenen Missbildungen Prävalenz bei Neugeborenen: 46 -50 %o n 25 -40 % der Säuglings- Mortalität n Faktor bei der Entstehung von Frühgeburten und Dystrophie n Schwere Zustände n Belastung: Kind, Familie, Gesellschaft n
Anzahl der Chromosomen aberrationen n 1970: ¨ n ~ 25 Krankheiten waren bekannt Heute: ¨~ 700 sind bekannt
Klinische Ausprägung Schweregrad: Fehlbildungen und Dysmorphien n „Maior”: Schäden von wichtigen Organ – Funktionen, funktionelle Behinderungen n ¨ Isoliert, Einzeldefekte (GI Atresien, Fallot – Tetralogie) ¨ Multiple Defekte: zwei oder mehr Organe, System – Schädigungen, -Krankheiten
Dysmorphien (Minor Missbildungen) n Informative morphogenetische Varianten Normale Organ-Funktionen, ästhetische Veränderungen ohne pathologische Wirkung ¨ Kombinationen ¨ - Epicanthusfalten
n Epikanthus falten n Präauriculäre/auriculäre Hautanhängsel n Zahn des Neugeborenen
n Überzählige Mamillen (Polythelie) n Klinodaktylie
n Parzielle Syndaktylie n Brachymesophalangie (Verkürzung der Mittelphalanx des 5. Fingers)
n Polydaktylie
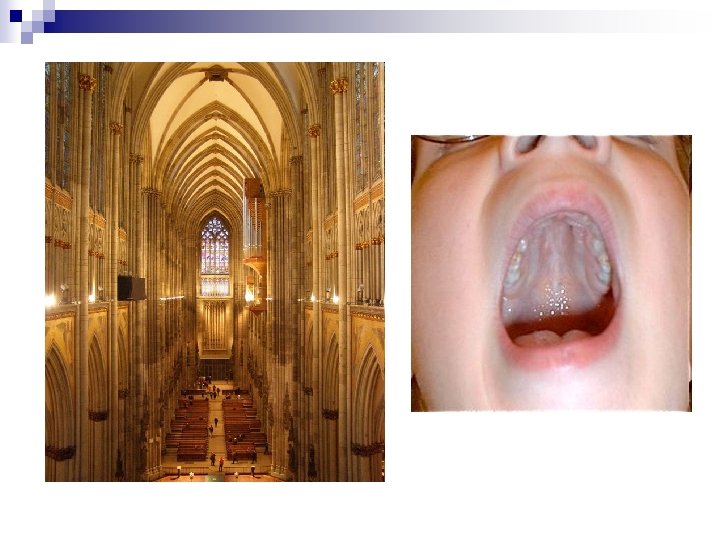
n Uvula bifida n Vierfingerfurche
Hypertelorismus (weiter Augenabstand)
Tiefsitzende (nach hinten rotierte) Ohren Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Verdacht auf eine Chromosomen- aberration Strukturelle Missbildungen des Schädels und des Gesichts (kraniofaziale Dysmorphie) Die mentale Entwicklung ist langsam oder retardiert Missbildungen und Symptomen mehrerer Organen Mütterliches Alter ist 35 Jahre oder mehr
Hauptbefunde der autosomalen Chromosomenaberrationen Wachstumsrückstand n Angeborene, multiple Fehlbildungen n Dysmorphiezeichen (Minor) n Verzögerung der geistigen und motorischen Entwicklung n
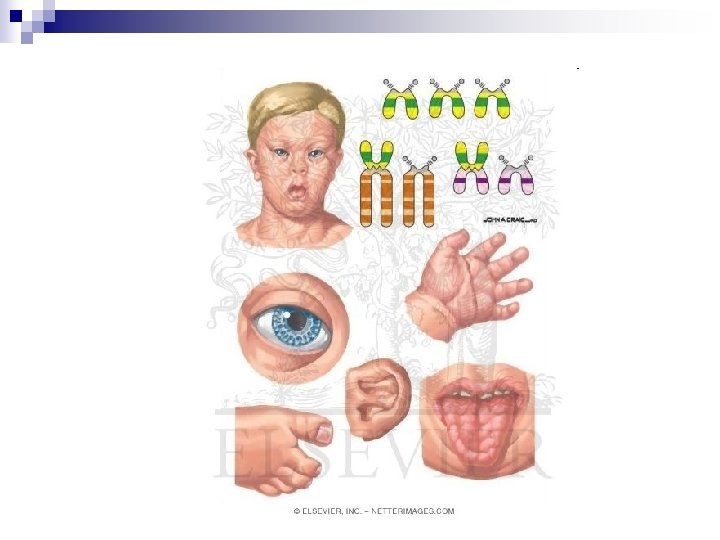
Trisomie 21 John Langdon Haydon Down (1828 -1896) England, Earlswood Asylum n Down- Syndrom n 21 q 22 Down’s Veröffentlichung: 1866 n
Trisomie 21 1: 700 - 1000 n Häufigste chromosomale Veränderung n n Klinische Symptome: Mikro-, Brachyzephalie, rundes Gesicht Kurzer / breiter Hals (Nackenödem) Flache Nasenwurzel Kleine, tief sitzende Ohren
Trisomie 21 Nach aussen ansteigende Lidachse, Epikanthus , Hypertelorismus n Offener Mund wegen schlaffem Muskeltonus n Grosse Zunge (Makroglossie) n Vierfingerfurche n Hoher, schmaler („gotischer”) Gaumen n
Trisomie 21 • • • Angeborene Herzfehler (50%) ( Atrio- Ventrikular- Kanal, Fallot - Tetralogie, Ventrikel- / Antrumseptumdefekt Gastrointestinale Fehlbildungen: Pankreas anulare, Duodenalatresie Katarakt, Myopie Sandalenlücke (grösserer Abstand zwischen Grosser und der 2. Zehe) Hypotonie und Überstreckbarkeit der Gelenke
Trisomie 21 Brushfield-Spots (Ablagerungen in Augen) Präsenile Demenz, Alzheimer • Mentalretardierung Kleinwuchs Häufige Infekte Erhöhtes Leukämierisiko Hypothyreose
Ursachen Freie Trisomie 21: vorwiegend mütterliche Ursache, meiotische Nondisjunktion n Translokation (Robertson): akrozentrische Chromosomen, 75% neu , 25% familiär n Mosaik-Trisomie n
Differentialdiagnose - Makroglossie Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Differentialdiagnose - Makroglossie Down – Syndrom n AD erbliche Makroglossie (MKNo. : 153630) n Hemihypertrophie n Glykogenose Typ II (Pompe) n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Hemihypertrophie Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Beckwith – Wiedemann (Exomphalos. Macroglossia-Gigantism)- Syndrom John Bruce Beckwith (1933 - ), Pä P diatr. Pathologe, USA, Hans Rudolf Wiedemann (1915 -2006), Deutscher Kinderarzt n AD, 11 p 15. 5 Deletion oder Duplikation n Prof. Wiedemann hat 12 Syndrome beschrieben Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Beckwith - Wiedemann - Syndrom Bei 3 oder mehr der folgenden Kriterien: n Makrosomie, Makroglossie n Viszeromegalie, Bauchwanddefekt: Omphalozele, Nabelhernie, Diastasis recti n Hemihypertrophie n Embryonale Tumorerkrankungen (Wilms- Tumor) Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Omphalozele Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Beckwith- Wiedemann - Syndrom n n n Adrenokortikale Zytomegalie Ohrfehlbildungen Nierenabnormalitäten Neonatale Hypoglykämie Gaumenspalten Sporadisch, selten familiäres Vorkommen Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Pätau - Syndrom (Trisomie 13) Klaus Pätau (1908 – 1975), Humangenetiker, Deutschland, USA, 1: 6000 47, XX, +13 bzw. 47, XY, +13 Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Pätau - Syndrom Psychomotorische Retardierung n Skalp-Defekte n Postaxiale ( auf der kleinfingerseite) Hexadaktylie n Herzfehler n Zystennieren n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Postaxiale Hexadaktylie Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Edwards Syndrom (Trisomie 18) John Hilton Edwards (1928 -2007) England, Humangenetiker Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Trisomie 18 n n Mikrognathie Kamptodaktylie der Finger und Zehen Klumpffuss Schwere Gedeihstörung Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Kamptodaktylie (fixierte Flexion) Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Ullrich – Turner - Syndrom Otto Ullrich (1894 -1957) Deutscher Kinderarzt, Henry Hubert Turner (1892 -1970), USA, Endokrinologe Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Ullrich -Turner - Syndrom (45, X): Lymphödem / Hände, Füsse, Pterygium colli Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Klinefelter Syndrom Harry Fitch Klinefelter (1912 - 1990), USA, Endokrinologe n Keimdrüsenunterfunktion im Pubertäts. Alter n 1– 2 von 1000 männlichen Neugeborenen Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Mikrodeletionen Analyse durch fluoreszierende In-situ. Hybridisierug (FISH) n Einige Mikrodeletionen schliessen dominante und geschlechtsgebundene Gene ein n Phänotyp eines oder mehrerer dominanter Erkrankungen („contiguous gene syndromes”) n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Mikrodeletions - Syndrome Wolf-Hirschhorn n Cri du chat n Williams - Beuren n WAGR n Prader-Willi/Angelman n Smith-Magenis n Miller-Dieker n Di. George/VCFS n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika 4 p 16. 3 5 p 15. 2/15. 3 7 q 11. 23 11 p 13 15 q 11 -12 17 p 11. 2 17 p 13 22 q 11. 2
Wolf – Hirschhorn – Syndrom Ulrich Wolf (1933 - ), Deutscher Humangenetiker, Kurt Hirschhorn (1926 - ), Humangenetiker , Österreich / USA Partielle Monosomie 4 p/4 p-Syndrom (4 p 16. 3) n 1: 50000 n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Wolf – Hirschhorn - Syndrom Dolichozephalie, hoher Stirn „griechischer Helm” Mentalretardierung Gedeihstörung Hypertelorismus Daumenhypoplasie n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Wolf - Hirschhorn - Syndrom Iriskolobom n Breite Nase n Lippen-Kiefer Gaumen-Spalte n Herzfehler n Urogenitalfehlbildungen n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Prader –Labhart – Willi - Syndrom Andrea Prader (1919 -2001), Schweiz, Kinderarzt u. Endokrinologe, Alexis Labhart (1916 - )Schweiz, Internist, Heinrich Willi (19001971), Schweiz, Kinderarzt Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Prader – Labhart -Willi - Syndrom 1 : 10000 – 20000 n Muskelhypotonie n Gedeihstörung n Ab dem Alter von 2 -3 Jahren Hyperphagie n Obesität n Minderwuchs n Geistige Behinderung n Hypogonadotroper Hypogonadismus
Prader – Labhart - Willi - Syndrom Hypogenitalismus, Kryptorchismus bei Knaben n Kleine Hände und Füsse n Hypopigmentierung n Diabetes mellitus n Herzinsuffizienz n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Prader- Labhart - Willi - Syndrom Ursachen n Paternale Deletion 15 q 11. 2 – q 12 (70%) n Gen: SNRPN – Small-nuclear. Ribonucleoprotein- Polypeptide- N n Alternative splicing Mütterliche uniparentale Disomie (UPD)(30%) n Imprinting- Fehler (<1%) n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Angelman Syndrom Harry Angelman (1915 - 1996), England, Kinderarzt Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Angelman - Syndrom „Happy Puppet syndrome” n n Psychomotorische Retardierung fehlender Spracherwerb bei besserem Sprachverständnis Ataxie Häufiges grundloses Lachen/Lachanfälle, leicht erregbar, mit Winken, Händeklatschen Krampfanfälle und EEG-Veränderungen, Mikrozephalie Grosser Mund, weiter Zahnabstand Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Di. George - Syndrom Angelo Mari Di. George 1921 -2009, Kinderarzt, USA n Velo- cardio- facialis (Shprintzen, Sedlackova, Mikrodeletion 22 q 11. 22)Syndrom Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Di. George/Shprintzen-Syndrom, VCFS, CATCH 22 n CATCH 22 = cardiac defect, abnormal facies, thymic hypoplasie, cleft palate, hypocalcemia n Aplasie /oder Hypoplasie des Thymus und der Parathyroidea Hypokalzämie (60%) Zellulärer Immundefekt Hypothyreose Konotrunkale (Aorta, Trunkus) Herzfehler (~75%) Dysmorphes Gesicht, Lippen-/Gaumen-/Kieferspalte Milde mentale Retardierung (~ 40%) n n n Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Fluoreszenz In - Situ Hibridisierung (FISH) Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
49, XXXXY
Vergleichende (komparative) genomische Hybridisierung (CGH) Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Bioinformatik Semmelweis Egyetem, II. Sz. Gyermekgyógyászati Klinika
Monogene Erkrankungen
Autosomal dominante Krankheiten Marfan – Syndrom Bindegewebsdefekt Prävalenz = 1 : 10000 – 20000 Klinische Symptome ◦ ◦ Skelett, kardiovaskuläres System, Augen: Linsenluxation, Bulbusverlängerung, Myopie, flache Kornea, Irishypoplasie
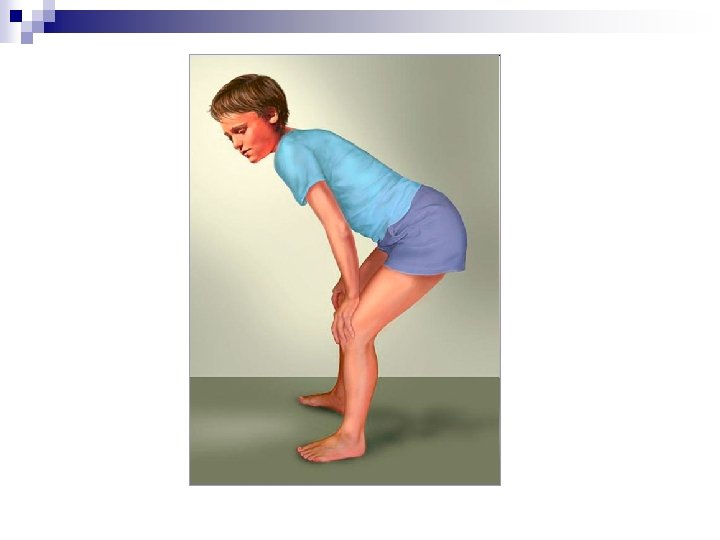
Marfan - Syndrom Dolichostenomelie Dysproportionierter Körper Thoraxdeformitäten (Trichterbrust, Hühnerbrust) Kyphoskoliose Senkfuss Hüftgelenksarthrose
Marfan - Syndrom Arachnodaktylie Fusszehen) (lange Finger +
Marfan - Syndrom Mittelklappenprolaps Aortendilatation, Aortenruptur
Niccolo Paganini Abraham Lincoln
Marfan - Syndrom FBN 1 – Gen (Fibrillin), 15 q 21. 1 Über 200 Mutationen, meist „Privatmutationen” Fibrillin: extrazelluläres Glykoprotein – Mikrofibrillen des Bindegewebes 25 -35%: Neumutationen Väterliches Alter ↑
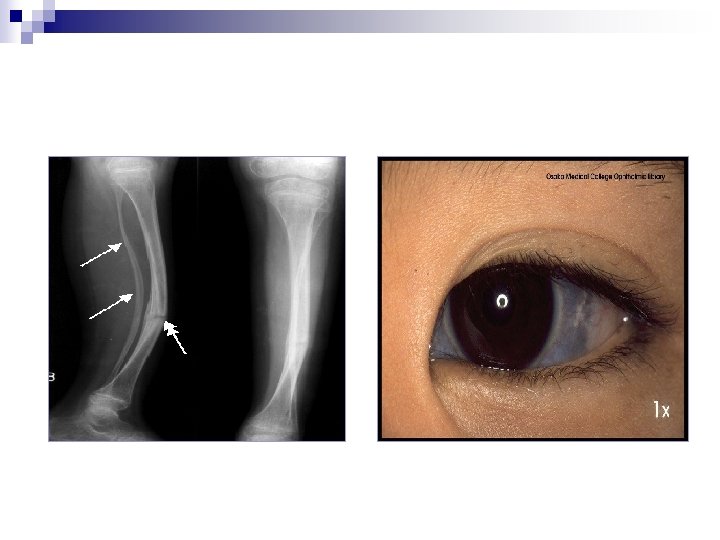
Osteogenesis imperfecta Klinische ◦ ◦ ◦ Symptome: Abnorme Knochenbrüchigkeit Blaue Skleren Otosklerose Schlaffheit des Bindegewebes und der Haut Überstreckbare Gelenke
Dentinogenesis imperfecta
Autosomal rezessive Krankheiten Mukoviszidose /Zystische Fibrose /CF Inzidenz = 1: 2500 Heterozygoten: 1/ 20 – 25 Störung des Chloridionentransportes Wenn jeder 20. ein heterozygoter Träger einer Mutation im CFTR – Gen ist, dann beide Eltern sind Träger mit einer Wahrscheinlichkeit von 1: 400. Das Risiko für das Kind: 1: 1600
CF Mekoniumileus / Neugeborenen Gedeihstörung/ Minderwuchs Chronische Bronchitis, Lungenentzündungen, Atelektasen, Bronchiektasen
CF Exokrine Pankreasinsuffizienz Männer: Aplasie der Vas deferens mit Infertilität (obstruktive Azoospermie) Idiopathische Pankreatitis Diagnose: Schweisstest (Na. Cl >70 mval/l) Mutationsanalyse
CF Cystic Fibrosis Transmembrane conductance Regulator (CFTR) - Gen Mehr als 2000 Mutationen , 7 q 31. 2 Häufigste Mutation: F 508 del (3 bp Deletion – Phenylalanin) Homozygoter Gendefekt Compound Heterozygotie Schwere und milde Mutationen Für F 508 del Mutation ein Nord – Süd - Gefälle
Mutation F 508 del
X –chromosomal rezessive Krankheiten Hämophilie A und B Muskeldystrophie Duchenne (DMD) Fehlen des Dystrophins Inzidenz= bei Jungen/ Männern 1: 3500 Klinische Symptome: ◦ Ca. im Alter von 3 -4 Jahren: Dystrophien der Beckengürtel, - Waden, - und Oberschenkelmuskulatur
DMD Pseudohypertrophie der Wadenmuskulatur Gower – Zeichen Erhöhte CK – Werte Herz: Reizleitungsstörung, dilatative Kardiomyopathie Lebenserwartung niedriger, Ateminsuffizienz
DMD Dystrophin – Gen, Xp 21. 2 (79 Exons)
Muskeldystrophie Becker (BMD) Milder als DMD, späteres Beginn Kein Fehlen , sondern Änderung der Struktur des Proteins Dystrophin
X- chromosomal dominante Krankheiten Vitamin – D- resistente hypophosphatämische Rachitis (Phosphatdiabetes) Rett – Syndrom Inzidenz = bei Mädchen 1: 10000 – 15000 Klinische Symptome: Nach den ersten 6 -8 Monaten Entwicklungsstillstand
Rett - Syndrom MECP 2 – Gen(methyl – Cp. G –binding protein -2), Xq 28 Frühe, atypische Form: CDKL 5 –Gen (cyclin-dependent kinase-like 5), Xp 22. 13 Kongenitale Form: FOXG 1 – Gen (forkhead box G 1), 14 q 12
Rett - Syndrom Verlust erworbener Fähigkeiten Stereotype Handbewegungen (Waschbewegungen) Geistige Retardierung Epileptische Anfälle Neurologische Symptome (Ataxie, autistische Symptome)