Investigatorinitiated Research What Are your Responsibilities Roxana Mehran
 and")
Investigator-initiated Research What Are your Responsibilities? Roxana Mehran, MD Professor of Medicine (Cardiology) and Health Evidence Policy Director of Interventional Cardiovascular Research and Clinical Trials The Icahn School of Medicine at Mount Sinai, New York, NY Chief Scientific Officer Cardiovascular Research Foundation, New York, NY Cardiovascular Research Technology - CRT Washington, DC

Disclosure Statement of Financial Interest Within the past 12 months, I or my spouse/partner have had a financial interest/arrangement or affiliation with the organization(s) listed below. Affiliation/Financial Relationship Company • Grant/Research Support • Astra Zeneca, BMS, The • Consulting Fees/Honoraria • Janssen (J+J) Medicines Company, Lilly/ DSI

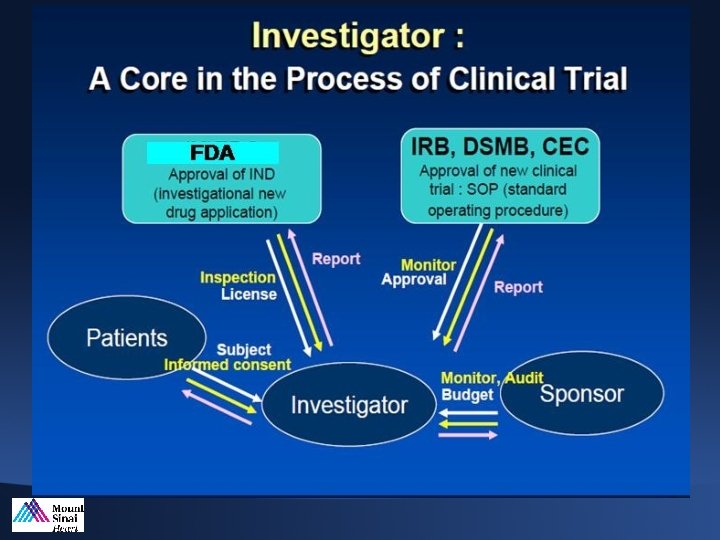
Investigator’s Responsibility • The investigator is responsible for insuring that an investigation is conducted according to the signed agreement, the investigational plan an applicable regulations for protecting the rights, safety and welfare of human subjects in the studies she/he conducts 21 CFR 320 -60, 21 CRF parts 50 and 56, 21 CFR 812 -100, ISO 14155, ICH E 6

Role and Responsibilities of a Principal Investigator Role as a Principal Investigator is dynamic, interacting with the organizational and clinical components of a trial: • Trial sponsor • Any additional trial committees (eg, Executive and/or Steering Committee) • Coordinating center • Regulatory agency • Site investigators and research staff

Role and Responsibilities of a Principal Investigator Planning and Initiation Protocol development Holder of IDE or IND applications* Preparation of budget and grant submission* Participation in regulatory presentations Site selection and recruitment Investigator and research staff training and protocol review (‘Investigator Meeting”) *Investigator-initiated trials

Sponsor • The sponsor takes responsibility for and initiates a clinical investigation. 21 CFR 312. 50 • The sponsor may be a device or pharmaceutical company, a private or academic organization, or an individual.

Sponsor-Investigator • A Sponsor-Investigator is an individual who both initiates and conducts a clinical investigation and under whose immediate direction the investigational device used or a drug is being administered or dispensed.

Caveats • For administrative reasons, only one individual should be designated as sponsor. • If a pharmaceutical company supplies the drug, but does not submit the IND, the company is NOT the sponsor. • Contract with supplier should define who does what.

Overview of FDA Requirements for Sponsor-Investigators • Review the applicable federal regulations before performing any sponsor duties • If you are a Sponsor-Investigator, you MUST meet the requirements of both the sponsor and the Investigator. • http: //www. access. gop. gov/nara/cfr/wai sidx_00/21 cfr 312_00. html

If you still want to be a Sponsor-Investigator • The following needs to be in place BEFORE you apply for the IDE/ IND.

To Do List: Study Organization • • • Sponsor identification Funding PI & Team work Steering Committee Data safety Monitoring Board Clinical Events Committee Data Management/ Stats /CRF Core labs Site Selection Monitoring (external)
 Have good knowledge of and experience in the")
Resources Experienced teamwork (PI and staff) Have good knowledge of and experience in the field of study defined by the protocol Have the necessary resources to participate in and take full responsibility for the proper conduct of the study Infrastructure (stats, databases, IRB protocols, location) necessary facilities, including emergency equipment and appropriate medical, paramedical and clerical staff to support the study Funding access to the drug and or the device

Investigational Protocol • The investigator must have a good knowledge of the protocol, protocol related documents and the requirements of the local participant code of rights and privacy legislation • The protocol and related documents should be approved and signed by the principal investigator and a representative of the sponsor • A budget in the form of a written contract should be established and documented in the investigator's information package for each study • Data ownership should be stated clearly in the protocol or contract

IRB, Case Histories, Payments • The sponsor must: ¡ require investigators to meet local IRB requirements. ¡ maintain complete and accurate records of payments made to clinical investigators. ¡ require investigators to keep case histories on each individual administered the investigational drug or employed as a control in the investigation

Study Essentials Recruitment: • A key factor – no results without participants. • Balance between being too specific or too diffuse – risk of having too narrow inclusion/exclusion criteria versus introduction of confounders • It’s not entirely your study – best if participants are genuinely interested in the study

During The Study The PI is responsible for the collection, quality, recording, maintenance and retrieval of source data arising from the clinical study • Each CRF case book (and selected pages) must be signed and dated by the investigator, or designated person, then stored securely • The investigator should make the data available on a timely basis must be available for agreed visits by the monitor during the study and also cooperate in the data editing, quality control and audit also by FDA

Monitoring and Compliance • Will select a monitor to oversee the progress of the investigation. 21 CFR 312. 53(d) • Will comply with FDA regulations regarding emergency use of test article. 21 CFR 312. 54
 Organizational Flow DMC reviews data from: • • • Case")
Data Monitoring Committee (DMC) Organizational Flow DMC reviews data from: • • • Case Report Form (CRF) Adverse Event (AE) Central laboratory Any other sources Protocol violations Endpoints adjudication CRO=contract research organization Chow et al. J Biopharm Stat. 2012. 22(4)853 -867.

Monitoring and Termination • The Sponsor shall monitor the progress of all clinical investigations being conducted under their IND. 21 CFR 312. 56(a) • The Sponsor shall terminate investigator’s participation when investigators fail to follow protocol. 21 CFR 312. 56(b)

Drug Safety & Effectiveness • The Sponsor shall discontinue the study if the investigational drug presents an unreasonable and significant risk to subjects • The FDA, IRB, and all investigators must be notified of the discontinuance. 21 CFR 312. 56(d)

Reporting Requirements • Progress Reports – Your investigators must furnish these to you as the sponsor. 21 CFR 312. 64(a) • IND Safety Reports – Adverse Reactions 21 CFR 312. 32 • Annual Reports – Within 60 days of anniversary of IND 21 CFR 312. 33

Reporting Requirements • Final Reports – must obtain from your investigators and submit to FDA 21 CFR 312. 64(c) • Financial Disclosure Reports from investigators must be received and updated for one year post study completion. 21 CFR 312. 64(d)

Investigator Sponsor Clinical Study Summary: 1. 2. 3. 4. 5. 6. 7. 8. 9. Examine your own experience and resources to conduct the investigation Consult with experienced in the field share with them your investigational plan Consult with a regulatory expert. Contact the FDA to verify whether you need to submit an IDE/IND Make sure the protocol is adequate, hypothesis defined, statistically powered, feasible to conduct in a reasonable time Assemble your study team and partners Documentation is essential. Read all the regulation and your responsibilities and ask yourself weather you are still interested. Cover your bases with legal support usually provided by your institution

Investigator Sponsor Clinical Trials References: • Guidance for Clinical Trial Sponsors: Establishment and Operation of Clinical Trial Data Monitoring Committees US Food and Drug Administration (FDA) OMB Control No. 0910 -0581 (March 2006) • Chow et al. J Biopharm Stat. 2012. 22(4)853 -867. • http: //www. access. gop. gov/nara/cfr/wai sidx_00/21 cfr 312_00. html
- Slides: 25