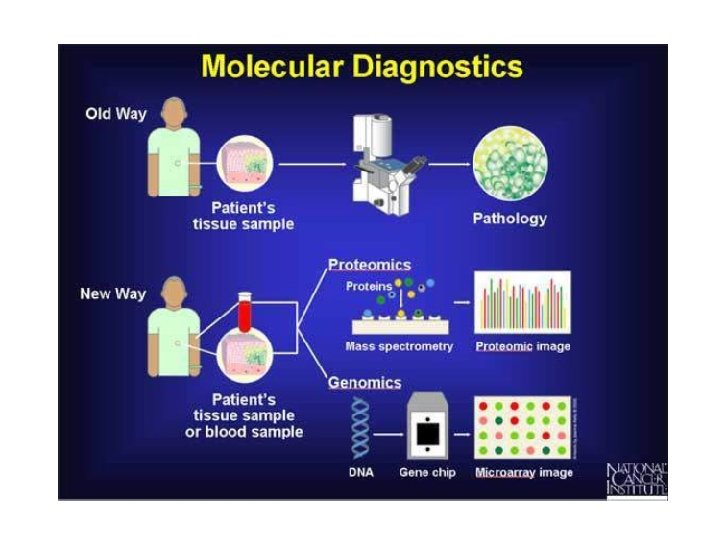
INTRODUZIONE ALLA DIAGNOSTICA MOLECOLARE Diagnostica molecolare n n






 §")
 § Il test diagnostico ideale è")










 viene estratto a) SANGUE totale")
. E’ composto da")

 Sacchetti trasporto campioni biologici c. pezzi chirurgici")









 da sangue in gradiente di Ficoll-Hypaque: miscela costituita da destrano ad")
 da sangue in gradiente di Ficoll La purificazione dell’RNA da sangue")

 e per favorirne la")
 2. Separazione dai contaminanti")




 per legare il DNA")


 46")










 + contaminazione reagenti Solo reagenti contaminanti")
 Gel")
")
 • Separazione di molecole")
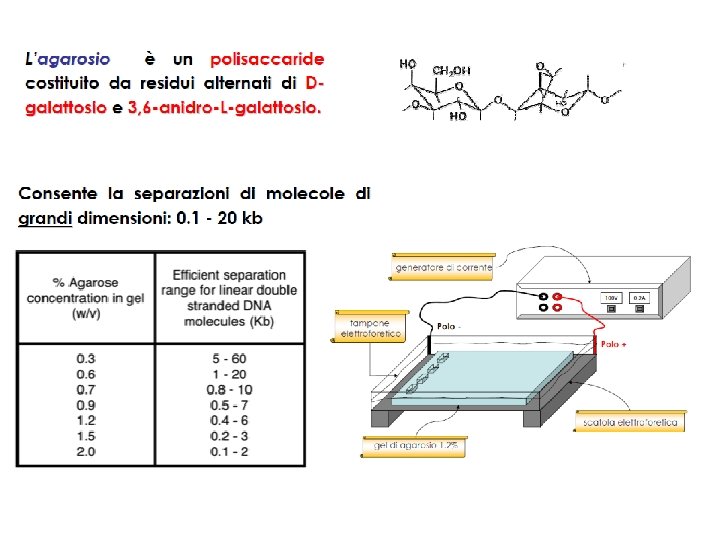

 65")
 • Elettroforesi per la separazione di frammenti di DNA")

- Slides: 67
INTRODUZIONE ALLA DIAGNOSTICA MOLECOLARE
Diagnostica molecolare n n n Branca di nuova acquisizione della medicina di laboratorio (biochimica clinica, ematologia, microbiologia, ecc. ); Si avvale di una serie di tecniche permettono di identificare lo stato patologico a partire dal meccanismo molecolare alla base delle malattie; In generale la diagnostica molecolare si occupa di: n n n Analizzare gli acidi nucleici per identificare alterazioni somatiche o ereditarie che sono causa di patologie o che predispongono all’insorgenza di malattie. Rilevare la presenza di acidi nucleici esogeni causa di infezioni da parte di agenti batterici o virus. Analizzare il DNA degli individui al fine di determinare la variabilità individuale (test di identità).
Lo sviluppo della diagnostica molecolare 1. Sviluppo delle tecniche del DNA ricombinante Comprensione a livello molecolare delle patologie 5. Progetto genoma 2. Identificazione di singoli geni (ibridizzazione molecolare) Identificazione di geni bersaglio a scopo diagnostico 3. Amplificazione del bersaglio (PCR) 4. Sistemi innovativi di rilevazione del segnale (automazione) Test molecolari
Campi di applicazione della diagnostica molecolare Le applicazioni diagnostiche molecolari sono numerose. Le discipline in cui hanno avuto maggior sviluppo sono: § Genetica molecolare § Diagnosi genotipica diretta § Diagnosi genotipica indiretta polimorfismi associati a malattie (RFLP) § Disordini del cariotipo (FISH) § Microbiologia § Diagnosi di agenti infettivi (batteri, virus, funghi) § Dosaggio dei livelli viremici. § Caratterizzazione genotipica del microrganismo (resistenza agli antibiotici). § Oncologia Molecolare § Identificazione di geni tumore specifici (oncogeni). § Valutazione di clonalità cellulare nei tumori ematopoietici § Medicina legale § Test di identità, paternità o maternità (DNA fingerprinting)
Principali tecniche molecolari in uso presso i laboratori di diagnostica § Tecniche di ibridizzazione-blotting § Ibridizzazione (Southern, Northern, Dot blot) § Tecniche di amplificazione § PCR, LCR, RT-PCR § PCR quantitativa § TMA, NASBA (amplificazione di RNA) § b. DNA (DNA ramificato) n n Sequenziamento di DNA Citofluorimetria Microarray Strumentazione di supporto: apparati per elettroforesi, spettrofotometri, fluorimetri, luminometri, termociclatori, sequenziatori ecc.
VALIDITA’ ANALITICA DI UN’ANALISI DIAGNOSTICA E’ data dalla specificità e dalla sensibilità analitiche e rappresenta l’accuratezza del test nell’evidenziare l’alterazione genetica La validità clinica rappresenta invece l’accuratezza del test nel diagnosticare precocemente la malattia
I test molecolari offrono una specificità e sensibilità elevate § Elevata specificità (analitica) § Possibilità di discriminare acidi nucleici che differiscono anche di una singola base (sia DNA che RNA). § Elevata sensibilità (analitica) § Possibilità di determinare la presenza di un determinato acido nucleico anche se questo è in piccolissime quantità § Capacità di amplificare il bersaglio per poterlo rilevare. § Rapidità dei test. § In molti casi i saggi molecolari sono più rapidi rispetto ai tradizionali sistemi di diagnosi (es. identificazione dei microrganismi) § Possibilità di automazione (in rapido sviluppo)
Sensibilità e specificità dei test clinici (TEST PERFORMANCE) § Il test diagnostico ideale è una determinazione analitica che dà sempre un risultato POSITIVO se un individuo presenta un certo carattere (HA una certa malattia), ed è sempre NEGATIVO se non presenta quel carattere (NON HA la malattia) (Test perfetto) § Tuttavia nei test può accadere, seppur con bassissima percentuale, che soggetti malati vengono classificati come negativi (falsi negativi) e soggetti sani non riconosciuti come tali (falsi positivi)
Il valore di qualunque test clinico è direttamente proporzionale alla sua sensibilità diagnostica e alla sua specificità diagnostica La sensibilità diagnostica: è la misura della frequenza di positività a un test in presenza della patologia considerata. Sensibilità = VP VP + FN * 100 VP= veri positivi FN= falsi negativi La specificità diagnostica: è la misura della frequenza di negatività a un test in assenza della malattia Specificità = VN VN + FP * 100 VN= veri negativi FP= falsi positivi
La validità di un test diagnostico: è direttamente correlata a specificità e sensibilità. Più è elevata, più il test si può definire ideale Validità o attendibilità test diagnostico = (VP + VN) VP= veri positivi FN= falsi negativi
Significato clinico dei test molecolari § Diagnostico n § Prognostico n § Facilitano il monitoraggio del decorso e dello stato di avanzamento delle malattie Terapeutico n § Permettono una diagnosi precoce di molte patologie Sono in grado di aiutare la scelta della terapia da adottare (farmacogenetica) Predittivo n Consentono di ottenere informazioni anche di tipo predittivo ossia danno indicazioni sui rischi futuri degli individui di sviluppare malattie (es. analisi prenatale).
Limiti attuali dei test molecolari v Possono essere utilizzati solo se è conosciuta la patogenesi molecolare della malattia: ¨ Non facilmente utilizzabili nelle patologie multigeniche o che coinvolgono anche fattori ambientali (diabete, aterosclerosi, malattie metaboliche, alcuni tipi di tumore, ecc. ). v Alcuni test sono ancora laboriosi o necessitano di attrezzature complesse v I costi delle analisi sono talvolta elevati v Non tutte le metodiche basate su test molecolari sono state ancora validate
1. RACCOLTA DEI CAMPIONI, MANIPOLAZIONE, PREPARAZIONE E PROCESSAMENTO
Gli esiti degli esami di laboratorio influenzano almeno il 70% delle decisioni cliniche (Silverstein, 2003) Laboratori Clinici: lo Standard Internazionale ISO 15189: 2007 (aggiornato 2012) regola il sistema di gestione dei laboratori di diagnostica -> personale, strutture, trattamento e utilizzo dei campioni -> controllo dell’intero processo (dalla richiesta di esami, all’identificazione del paziente, fino alla produzione di un referto nel quale i risultati sono “trasformati” in informazioni utili per la diagnosi del paziente)
ISO 15189 : 2003 “I servizi di Medicina di Laboratorio sono essenziali per la cura dei pazienti e devono, quindi, essere in grado di soddisfare i bisogni di tutti i pazienti e del personale clinico responsabile delle cure”. Tali servizi comprendono 1. la formulazione delle richieste, 2. la preparazione dei pazienti, 3. l’identificazione dei pazienti (dati anagrafici, anamnesi), 4. la raccolta dei campioni, 5. il trasporto, 6. la conservazione, 7. la manipolazione e l’analisi dei campioni clinici, accanto alla 8. validazione, 9. all’interpretazione e alla refertazione degli stessi e in aggiunta alle considerazioni sulla sicurezza e sull’etica dei processi caratteristici del Laboratorio Clinico“
Il test di laboratorio è in grado di produrre informazioni importanti per la clinica. Però oltre al test devono essere prese in considerazione una serie di variabili, pre- e post analitiche capaci di garantire i risultati comunicati nel referto.
Dal campione biologico al referto: • Richiesta • Prelievo • Accettazione • Programmazione dell’iter diagnostico • Indagini di laboratorio • Interpretazione del risultato • Refertazione • Consulenza genetica (se necessario)
Le tre fasi dell’analisi di un campione v Fase preanalitica (la variabilità è legata al paziente o alla conservazione del campione) § v v Raccolta, trasporto (o spedizione), accettazione, smistamento e conservazione dei campioni Fase analitica (la variabilità è legata al metodo di analisi) § Preparazione del campione: § estrazione dell’acido nucleico § verifica della qualità dell’acido nucleico purificato § conservazione del campione di acido nucleico § Esecuzione dell’analisi § Acquisizione del dato e interpretazione del risultato. Fase postanalitica § Refertazione, archiviazione o smaltimento dei campioni.
FASE PRE-ANALITICA Campioni biologici da cui il DNA (RNA) viene estratto a) SANGUE totale fresco o congelato (anticoagulanti: EDTA o sodio citrato, evitare eparina). SIERO o PLASMA (ricerca di DNA virale, anticorpi, ecc. ) b) Altri LIQUIDI CORPOREI (espettorato bronchiale, liquidi cavitari, liquido cefalo-rachidiano, urine, ecc) c) CAMPIONI TISSUTALI (biopsie) § § § Freschi Congelati (azoto liquido poi -80°C) Immersi in paraffina d) CELLULE IN COLTURA (es. villi coriali, cellule fetali da amniocentesi) e) Tracce di DNA da campioni vari (tamponi boccali, capelli, macchie di sangue, liquido seminale, mozziconi di sigaretta ecc. )
Plasma = componente liquida del sangue (non comprendente la parte cellulare). E’ composto da acqua, proteine, glucosio, aminoacidi, lipidi, ormoni, urea e urati, ioni, anidride carbonica, ossigeno, vitamine Siero = è costituito dal plasma (parte liquida del sangue) privato del fibrinogeno, del fattore VIII, del fattore V e della protrombina. Si può isolare da un prelievo del sangue dopo centrifugazione (la parte corpuscolata si separa dalla fase liquida del sangue) Buffy coat = componente del sangue che contiene globuli bianchi e piastrine
SANGUE INTERO costituito da - parte corpuscolata - plasma Si mantiene rendendo il sangue incoagulabile con anticoagulanti PLASMA sangue privato degli elementi corpuscolati Si ottiene da sangue prelevato con anticoagulante separandolo dagli elementi corpuscolati mediante centrifugazione a 2. 000 rpm per 10 -15 min. SIERO plasma privato del fibrinogeno Si ottiene dal sangue intero senza aggiunta di anticoagulanti • coagulazione (2 ore a t° ambiente) –retrazione del coagulo • separazione per centrifugazione (2. 000 rpm per 10 -15 min. )
inclusione biopsie in paraffina c. urine (urinocoltura) Sacchetti trasporto campioni biologici c. pezzi chirurgici (formaldeide)
FASE PRE-ANALITICA Raccolta e conservazione dei campioni biologici La correttezza del risultato analitico dipende dal mantenimento delle peculiarità chimiche e biologiche del campione stesso. La qualità più elevata di DNA/RNA si ottiene da MATERIALE FRESCO. Se ciò non è possibile i campioni vanno conservati a basse temperature. Campioni per DNA: Conservare i campioni biologici a 2– 8 °C per 24 -48 ore. Vanno congelati a – 20/-80 °C periodi più lunghi. Non congelare e scongelare i campioni più volte: causa frammentazione del DNA. Campioni per RNA: Conservare i campioni biologici a -80 °C anche per lunghi periodi. Meglio impiegare anche stabilizzanti che contengono inibitori dell’enzima RNasi
Preparazione dei vari tipi di campione prima dell’estrazione degli acidi nucleici • Sangue: prelevato e messo in provette con anti-coagulante. Può essere usato come tale per isolare il DNA. Per isolare l’RNA è preferibile separare le cellule mononucleate del sangue (linfociti, monociti, granulociti) ed estrarlo da queste L’anti-coagulante eparina e l’emoglobina disturbano l’attività di enzimi quali DNA polimerasi e trascrittasi inversa-> preferire EDTA o sodio citrato come anticoagulanti; effettuare la lisi degli eritrociti • Urina, liquidi cavitari: centrifugazione per separare la componente cellulare dalla frazione liquida
• Liquor: concentrazione per centrifugazione su filtri con porosità definita per ottenere molecole con PM>100 Dalton • Campioni delle vie respiratorie (espettorato, lavaggio bronco-alveolare): fluidificazione con enzimi mucolitici, concentrazione delle cellule per centrifugazione • Campioni da biopsie: disgregazione del tessuto (omogenizzazione) e lisi delle emazie eventualmente presenti • Villi coriali: villocentesi entro 10 -13 settimane di gestazione. Diagnosi anomalie cromosomiche e genetiche dopo 5 giorni dal prelievo a partire da colture cellulari dei villi (distrofia muscolare, fibrosi cistica, talassemie, emofilie, corea di Huntington, ecc. ).
• Liquido amniotico: prelievo entro 15 -17 settimane di gestazione, cellule di origine fetale vengono messe in coltura per i test diagnostici
FASE ANALITICA Sistemi di estrazione degli acidi nucleici. Obiettivo: ottenere campioni di DNA/RNA privi di molecole contaminanti che possono alterare od inibire le successive fasi dell’analisi. Numerosi sono i metodi per isolare il DNA/RNA. Principi generali per la purificazione degli acidi nucleici sono: n Lisi delle cellule (o dei batteri) e rilascio del contenuto. n Separazione del DNA/RNA dal contenuto cellulare in particolare dalle proteine (enzimi) contaminanti. n Recupero (e concentrazione) del DNA/RNA purificato. Il recupero deve selezionare uno solo dei due acidi nucleici.
Many different methods and technologies are available for the isolation of genomic DNA. All methods involve: Ø A. disruption and lyses of the starting material followed by Ø B. Removal of proteins and other contaminants and finally Ø C. Recovery of the DNA
Parametri da tenere in considerazione per la scelta del metodo: 1. Tipo di campione (batteri, virus, tessuti, sangue ecc. ) 2. Grado di purezza (es. per digestione enzimatica, amplificazione, ecc. ) 3. Integrità del DNA (se serve DNA ad alto peso molecolare) o RNA 4. Velocità del processo, facilità d’uso (numero di campioni da analizzare). 5. Costo della preparazione
Principi generali per l’isolamento del DNA genomico 1. La lisi delle cellule (cellule eucarioti , tessuti, cellule procarioti -> pareti) è un passo obbligato per tutte le metodiche di estrazione. Si ottiene con soluzioni che contengono: 1. Un detergente che dissolve le membrane (es. SDS: sodio dodecil solfato, NP 40, ecc. ) 2. Una proteasi che degrada le componenti proteiche = deproteinizzazione (es. proteinasi K). 3. Spesso è aggiunto anche EDTA (acido etilendiamminotetracetico), chela gli ioni bivalenti necessari all’azione delle nucleasi endogene. Note: n n L’isolamento da batteri richiede la lisi enzimatica della parete ottenuta con lisozima. Isolamento da tessuti: è utile disgregare i tessuti con un omogenizzatore. n Se si vuol rimuovere l’RNA dalle soluzione di lisi sono aggiunte RNAsi e viceversa n Per estrarre DNA da sangue intero (non separato nelle sue componenti) si devono lisare le emazie e eliminare Hb per centrifugazione. Per RNA da sangue si devono per forza isolare
omogenizzatori
Separazione linfo-monociti (PBMC) da sangue in gradiente di Ficoll-Hypaque: miscela costituita da destrano ad alto peso molecolare e da sodio metrizoato. 1) destrano -> induce pseudoagglutinazione delle emazie per modificazione della carica elettrica di qs cellule 2) Na metrizoato -> consente di ottenere soluzioni ad alta densità
Separazione linfo-monociti (PBMC) da sangue in gradiente di Ficoll La purificazione dell’RNA da sangue richiede la separazione della componente cellulare nucleata che sarà sottoposta poi a estrazione. I neutrofili (granulociti ) degranulano facilmente e liberano enzimi che possono facilitare la degradazione dell’RNA o di un’eventuale componente virale
- Monocytes will be isolated by utilizing their ability to bind to plastic surfaces; - T and B lymphocytes will be separated using activated erythrocytes from sheep with specific antibodies to bind specific antigens on T and B-cells surface; - The different cell populations will finally be studied using flow cytometry
Deproteinizzazione Serve per evitare la degradazione degli acidi nucleici (enzimi) e per favorirne la purificazione
Principi generali di isolamento di campioni di DNA genomico (continua) 2. Separazione dai contaminanti (deproteinizzazione) e 3. recupero del DNA Metodi in fase liquida di estrazione del DNA: 1. Metodo del salting-in salting-out. 1. Aggiunta di soluzioni saline (Na. Cl) ipotoniche e ipertoniche in modo da far lisare le cellule e poi precipitare le proteine ed altri contaminanti 2. Precipitazione e recupero del DNA dopo aggiunta di alcoli quali isopropanolo (conc. finale 50%) o etanolo (conc. finale 70% ) e centrifugazione del campione. 2. Estrazione organica. 1. Estrazione organica con fenolo/cloroformio o Trizol: permette di separare i componenti del lisato in una fase acquosa (contenente DNA e RNA) ed una fase organica (contenente proteine).
2. Precipitazione del DNA per aggiunta di sali (sodio acetato 0, 3 M, p. H 5. 2) e un alcool come isopropanolo o etanolo. Centrifugazione del campione. Fase finale comune: al termine di qualsiasi procedura di estrazione del DNA, si osserva il recupero del pellet di DNA e la sua risospensione in tampone TE (Tris 10 m. M EDTA 1 m. M p. H 7. 5) o in acqua ultrapura N. B. In questo tipo di estrazione il fenolo serve per denaturare le proteine mentre il cloroformio solubilizza i lipidi
Estrazioni con fenolo-cloroformio Il p. H del fenolo influenza la separazione dei due acidi nucleici
ESTRAZIONE SALTING IN-SALTING OUT
Metodi in fase solida di estrazione del DNA-> kit commerciali La separazione del DNA dagli altri componenti cellulari può avvenire anche in fase solida. Principali passaggi: § Lisi delle cellule (vedi protocolli in fase liquida) § Passaggio in una resina insolubile (resina di silice) che lega in maniera specifica il DNA (e/o l’RNA) – adsorbimento - § Lavaggio della resina con tamponi che permettono la separazione dai contaminanti. Dopo aggiunta di etanolo 70% § Eluizione del DNA (o dell’RNA) dalla resina con acqua o soluzioni a bassa concentrazione di sali (TE) che ne facilitano il distacco
In alternativa: resine a scambio anionico con gruppi DEAE (dietilaminoetil) per legare il DNA (eluizione con sali e precipitazione). Procedure in fase solida: veloci, facili da usare, possono essere adoperate con sistemi automatizzati. cell lysis
Sistema di estrazione automatizzato con utilizzo di sferette magnetiche 1. Lisi delle cellule (sangue, plasma, siero, tessuti) 2. Trasferimento in colonnine contenenti biglie di vetro trattate (magnetiche perché fatte di ossido di Fe o silice accoppiata a magnetite) a cui si legano DNA o RNA 3. Avvicinamento a un magnete che richiama sulla parete delle colonnine le biglie legate al DNA/RNA 4. Apertura dell’estremità inferiore della colonna per poter allontanare i contaminanti. Aggiunta di tamponi e di DNAsi o RNAsi (mentre il DNA o l’RNA sono trattenuti) DNA purificato 5. Allontanamento del magnete ed eluizione del DNA (purificato) dalla colonna con TE o acqua (le particelle magnetiche sono trattenute nella colonna).
Sistema di estrazione automatizzato con utilizzo di sferette magnetiche
Estrattore automatico DNA (per kit con biglie magnetiche) 46
Overview of DNA Extraction Break down the cell wall and membranes Centrifuge to separate the solids from the dissolved DNA Precipitate the DNA using isopropanol Conservazione del DNA: in tampone TE o acqua a 4°C per settimane oppure da -20°C a -80 °C per tempi più lunghi. Dissolve DNA Wash the DNA pellet with Ethanol and dry the pellet Centrifuge to separate the DNA from the dissolved salts and sugars
Estrazione organica vs Estrazione in fase solida: tempo impiegato, costi, reagenti impiegati DNA binding Washes with buffer Elution 30 -60 minutes 2 days 48
Estrazione automatizzata vs Estrazione manuale: tempo impiegato, costi, reagenti impiegati • Riduzione dei tempi per l’estrazione • Riduzione degli errori legati all’operatore • Capacità di processare da 6 a 96 campioni in 2 - 3 ore • Possibilità di partire da vari tipi di materiale biologico e alla fine di aliquotare il DNA o l’RNA estratto riducendo i rischi di contaminazione • Costo più elevato (acquisto estratto automatico + reagenti dedicati)
Estrazione dell’RNA totale Particolarità nei protocolli di estrazione dell’RNA: n L’estrazione dei campioni deve avvenire in tamponi di lisi contenenti inibitori delle RNAsi (guanidinio tiocianato o cloridrato). n Per evitare contaminazioni di DNA genomico nella preparazione dell’RNA, ai tamponi di lisi possono essere aggiunte DNAsi. n Per estrarre l’RNA virale si parte da siero o plasma (sangue senza componente cellulare) o fluidi corporei n L’RNA è una molecola che viene facilmente degradata dalle ribonucleasi (RNAsi), per minimizzare la loro azione occorre: n n Prestare attenzione a non introdurre RNAsi esogene (indossare guanti, evitare contatto superf. ). Utilizzare solo soluzioni e materiali sterili o trattati con DEPC (dietilpirocarbonato) n Conservare i campioni di RNA in ghiaccio durante la loro manipolazione. n Utilizzare per la manipolazione dell’RNA materiali dedicati (provette, puntali, filtri etc) tutti Rnasi free n Conservare l’RNA estratto a -80°C!!!
Rnasi e Dnasi sono Nucleasi Si trovano nelle cellule e, quindi, anche sulla pelle per cui si devono evitare i contatti diretti o indiretti tra mani e acidi nucleici Le Dnasi (enzimi che degradano il DNA): • per attivarsi richiedono ioni metallici • sono termolabili (autoclave) • sono facilmente inattivabili da agenti chelanti Le Rnasi (enzimi che degradano l’RNA): • non richiedono cofattori • possono adsorbirsi a vetro e plastica e rimanere attive • resistono alla sterilizzazione in autoclave
Analisi degli acidi nucleici La qualità e la quantità del DNA o dell’RNA ottenuto possono essere determinati attraverso metodi analitici non selettivi o selettivi • Non selettivi: non danno informazioni su specifiche sequenze • Spettrofotometria UV • Elettroforesi su gel di agarosio • Selettivi: danno informazioni sulla presenza /assenza nel DNA/RNA analizzato di specifiche sequenze o regioni • Dot blot/southern , northern blot, PCR
Quantificazione degli acidi nucleici La concentrazione dell’acido nucleico nel campione può essere stimata misurando l’assorbanza della soluzione del DNA nell’ultravioletto (legge Lambert-Beer A=ex. Cx. L) DNA A =260 nm = 1. 0 DNA=50 g/ml (se c’è RNA contaminante il valore sarà sovrastimato) Tenere conto di un eventuale fattore di diluizione se si misura in cuvette o in piastre da 96 di quarzo! Il grado di purezza della preparazione di DNA può essere valutato dal rapporto: A 260/A 280 > 1. 8 DNA è sufficientemente puro da proteine contaminanti A 260/A 230 = 2. 0 -2. 2 DNA è sufficientemente puro da reagenti contaminanti (fenolo, etanolo, ecc. ) RNA A =260 nm =1. 0 RNA = 40 g/ml (se c’è DNA contaminante il valore verrà sovrastimato) Tenere conto di un eventuale fattore di diluizione se si misura in cuvette o in piastre da 96 di quarzo! Il grado di purezza della preparazione di RNA può essere valutato dal rapporto: A 260/A 280 = 1. 8 -2. 2 RNA è sufficientemente puro da proteine contaminanti A 260/A 230 = 2. 0 -2. 2 RNA è sufficientemente puro da reagenti contaminanti (fenolo, etanolo, ecc. )
A=ex. Cx. L
Spettrofotometro classico
Spettrofotometro Nano. Drop
DNA (RNA) + contaminazione reagenti Solo reagenti contaminanti
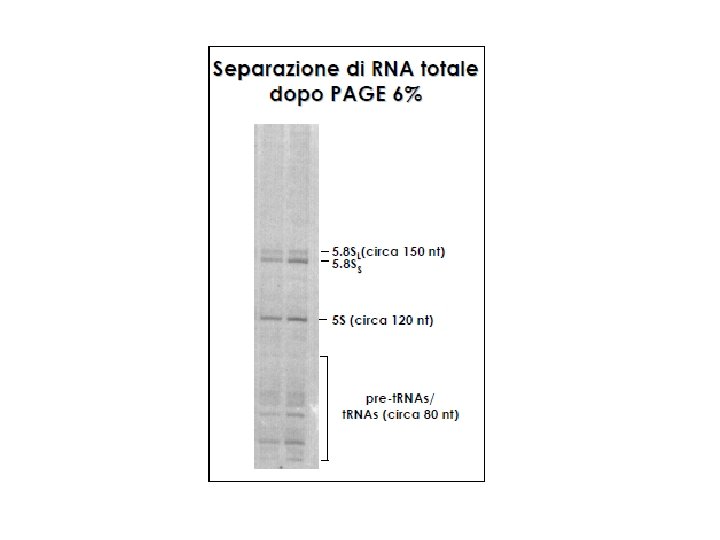
Analisi della qualità del DNA e dell’RNA mediante elettroforesi su gel d’agarosio (1%-2%) Gel di agarosio denaturante contenente formaldeide DNA r. RNA 28 S r. RNA 18 S 100 50 25 ng DNA genomico RNA degradato 5 S r. RNA, t. RNA, e altri piccoli RNA A =260 nm =1, 0 DNA=50 g/ml (se c’è RNA contaminante la concentrazione verrà sovrastimata) A =260 nm =1, 0 RNA=40 g/ml (se c’è DNA contaminante la concentrazione verrà sovrastimata)
Valutazione in corsa elettroforetica degli acidi nucleici (pulse-field gel electrophoresis)
Analisi di DNA e RNA mediante elettroforesi su poliacrilammide (PAGE) • Separazione di molecole di acido nucleico di dimensione inferiore a 1 kb • Discrimina molecole di DNA/RNA che differiscono per poche basi • Matrici da 6% al 18% (risoluzione 1000 -20 bp)
Analisi di DNA mediante elettroforesi su gel d’agarosio Gel di agarosio: le bande di DNA sono state messe in evidenza introducendo nel gel l’etidio bromuro (o il gel Red) intercalante che si inserisce tra le basi del DNA diventando fluorescente agli U. V. DNA fortemente degradato Frammenti di DNA di lunghezza nota (marker) DNA genomico da sangue intero umano http: //www. sumanasinc. com/webcontent/animations/content/gelelectrophoresis. html 64
Visualizzazione bande DNA gel PAGE con Etidio Bromuro (U. V. ) 65
Elettroforesi su campo pulsato (PFGE) • Elettroforesi per la separazione di frammenti di DNA genomico molto grandi (50 -250 kb) corrispondenti ai frammenti di digestione derivanti da cromosomi • Si impiega per caratterizzare ceppi batterici quando le analisi di tipizzazione non danno una risposta chiara • a) Digestione del DNA con E. R. b) produzione di frammenti di chilo o megabasi separati in gel sottoposto a un campo elettrico a direzione variabile -> le molecole di DNA avanzano e arretrano più volte separandosi nel tempo c) richiede -> switch campo elettrico, controllo temperatura, 24 -36 ore di corsa elettroforetica d) il pattern di bande risultante è caratteristico per ogni tipo di microorganismo (comparazione con corsa E. F. di controlli noti)
• L'azione del primo campo elettrico causa uno stiramento lungo il piano orizzontale delle molecole e il loro movimento all'interno del gel. • L'interruzione di questo campo e l'applicazione del secondo campo elettrico fa sì che le molecole si muovano nella nuova direzione. PFGE • In questo modo, variando continuamente la direzione del campo, si separano le molecole più piccole da quelle più grandi Analisi dei cluster elettroforetici del DNA di ceppi diversi di Escherichia coli in PFGE