Introduction to PCR and q PCR Part II




 properties Primers should have • 18 -24 bases • 40 -60%")
 problem • Primers that interact are amplified by PCR. •")














: Threshold cycle reflects the cycle number at which the")




















")




- Slides: 57
Introduction to PCR and q. PCR Part II: PCR!! Dr. Chaim Wachtel
q. PCR technical workflow DNA Extraction Sampling q. PCR RNA Extraction DNase treatment Reverse Transcription Data Analysis
Primer design
Primer design – key to successful PCR • Good primer design saves time and money • Advanced applications require even more stringent primer design – Multiplex – Low abundance
Good primer (pair) properties Primers should have • 18 -24 bases • 40 -60% G/C • Balanced distribution of G/C and A/T bases • Tm that allows annealing at 55 -65°C • No internal secondary structures (hair-pins) Primer pairs should have • Similar melting temperatures, Tm , within 2 -3 °C • No significant complementarity (> 2 -3 bp) – particularly not in the 3’-ends
The primer dimer (PD) problem • Primers that interact are amplified by PCR. • PD formation competes with the designed PCR and can compromise the reaction efficiency. Sense ´ 3 Antisense Cycling. . . Sense c. Antisense Sense Antisense
Solution to the PD problem • Reduce the formation of PDs by – Good primer design (avoid 3’ complementarity) – Minimal annealing time – Good laboratory practice – Hot. Start – Touch. Down • Reduce the signal from PDs by – Measuring fluorescence above the Tm of the PDs – Use sequence-specific probe
Considerations • Avoid targets with secondary structure • Avoid pseudogenes • Avoid genomic contamination by designing primers to span intron-exon-junctions PCR primers introns exons
Links for designing primers • http: //www. tataa. com/ • http: //www. ncbi. nlm. nih. gov/BLAST/ • www. premierbiosoft. com/netprimer/netprlau nch/netprlaunch. html • www. ensembl. org • http: //www-genome. wi. mit. edu/cgibin/primer 3_www. cgi • http: //www. bioinfo. rpi. edu/applications/mfo ld/dna/form 1. cgi • Primer Design- Beacon Designer/Alelle. ID • Primer express 3 (AB)
Primer Express • Located on Software 1 • Easy to use • Not fool-proof, but none of them are…. .
Primer design-work flow No Find sequence Design Primers Check Primers NCBI or Ensembl Primer 3 or similar software Satisfactory? for desired parameters - Tm - amplicon size - secondary structure - complementarity - specificity Netprimer, BLAST and similar software Yes Run PCR …and gel electrophoresis to check specificity and functionality
Taq. Man Probe Design • • Amplicon size 70 -150 bp Tm of probe 68 -70 °C G/C content 30 -70% No G at the 5´end Avoid runs of identical nucleotides Avoid secondary structure Avoid complementarity with primers HPLC purification
Popular dyes and quenchers • • FAM JOE HEX TET VIC ROX Cy • DABCYL • TAMRA • Black Hole Quenchers
RT-PCR • Housekeeping genes – What are they – How do you choose • • • Standard curve Primer Dimer Melt curve Optimization Test samples Reference
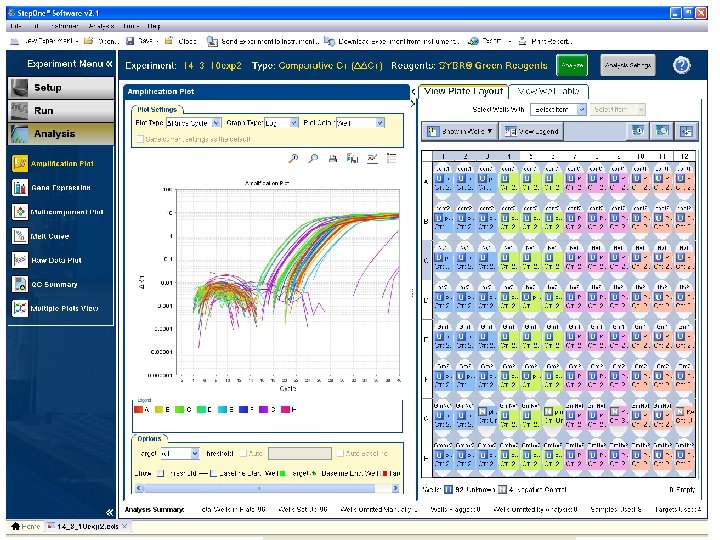
Workflow – preliminary data analysis Examine amplification curves Check/adjust baseline Check/adjust threshold Check your controls (NTC/positive controls/No-RT) Check your samples Perform experiment specific analysis
Baseline settings Baseline - is the initial cycles in PCR where there is little change in fluorecence signal, usually cycle ~315 • Set the baseline • Fixed number of cycles • Adaptive baseline • Control baselines in the linear scale (y-axis)
Raw data
Baseline adjustment
The different phases Exponential growth phase Plateau phase Part of exponential growth phase where signal > background (noise) Samples must be compared in the exponential phase
Setting threshold Logarithmic scale Linear scale • Purpose: Find a level of fluorescence where samples can be compared • The theoretical cycle where a sample intersect the threshold is called Ct Log Threshold level Ct values
Setting threshold Ct (threshold cycle): Threshold cycle reflects the cycle number at which the fluorescence generated within a reaction crosses the threshold. It is inversely correlated to the logarithm of the initial copy number
Setting threshold • Several methods available for threshold setting – Standard deviation of the noise for the first few cycles – Second derivative maximum (SDM) – Best fit of standard curve (highest r 2) – Manual setting A two-fold difference in copy number should have one Ct difference no matter where threshold is set within the exponential phase
Dilution series and standard curves • Used to control the quality of your assays • Absolute quantification – Standards = Diluted templates of known concentration – Standard curve = Ct of each standard sample is plotted against the known concentration – Used to determine concentrations of unknown samples – Absolute quantification is dependent on the quality of the standard curve
Standard curve Comment: Always cover the whole range of sample concentrations.
Interpretation of the standard curve • Linear regression Y = ax + b a = slope that gives efficiency of PCR from 10– 1/a = 1 + efficiency b = # of cycles for detecting one molecule
30 25 20 15 10 5 0 1. E+03 1. E+04 1. E+05 1. E+06 1. E+07 Concentration (log scale) 1. E+08 1. E+09
Relative quantification • Often there is no good standard available • Compare amount with reference • Reference genes • Genomic DNA • Spike • Ribosomal RNA • Example – Expression of target gene is 10% of the expr. of housekeeping gene. – Same gene in other tissue, expression is 100%.
Comparing treatments Finding the change in expression of the target gene in the sample compared to the control (no treatment) as a ratio, using a reference gene
MIQUE Nomenclature • MIQUE - Minimum Information for Publication of Quantitative Real-Time PCR experiments Suggested nomenclature • • Reference genes not housekeeping genes Quantification not quantitation Hydrolysis probes not Taq. Man probes Quantification cycle Cq replaces Ct, Cp, TOP
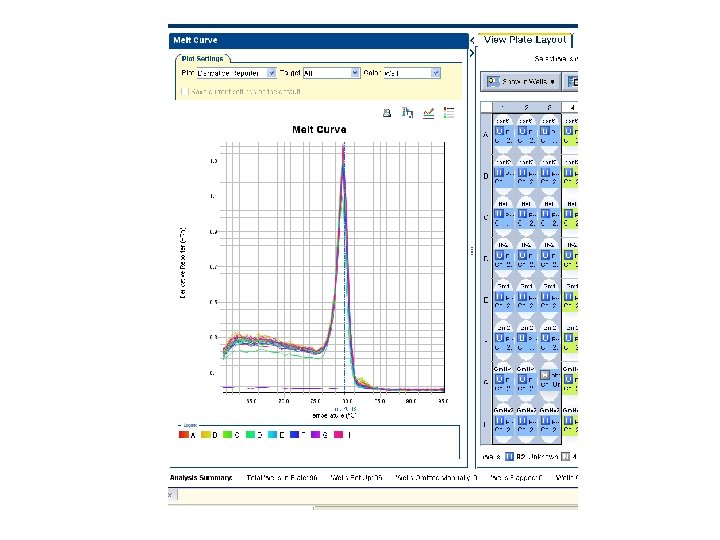
Melting curve analysis • Melting curves are obtained by measuring the fluorescence while increasing temperature • Use a dye binding to double stranded DNA 70 80 90 95 Temp
Melting curve analysis • Confirms formation of the expected product (each ds. DNA has its characteristic melting temp Tm) • Distinguishes between specific PCR products and non-specific products (e. g. primer-dimers) • High resolution melt – mutation and methylation analysis
Melting curve 1 st derivative Tm = 90 °C Melting temperature Tm is characteristic of the %GC, length and sequence. The product can be identified from the Tm. Tm = 81. 5 °C
4 -steps PCR can be used to eliminate primer-dimer signals 40 cycles
Example – 4 steps PCR
100% efficiency 75% efficiency
100% efficiency 90% efficiency
80% efficiency 50% efficiency
Reference primer efficiency 100% 90% 80% 75% 50% GMNv 1 3. 305 3. 823 4. 456 4. 826 7. 47 VN 1 3. 186 3. 451 3. 775 3. 924 4. 991 Ratio 1. 04 1. 11 1. 18 1. 23 1. 5
RT-PCR –testing samples • ALWAYS perform melt curve • ALWAYS run negative controls – No RT – No template • Always run standard curve • Triplicate of each sample!!
Requirements for RT-PCR Experiment • • • Always perform standard curve All samples in triplicate NTC control No RT control Prepare mix without c. DNA; add this to each tube separately Divide plate by gene and not sample Do not need reference gene on every plate Melt Curve Check RNA- otherwise don’t bother with experiment Do not rely on only 1 reference gene- check more than one per project • Every project is different! • Don’t be afraid to ask me questions, especially BEFORE starting the project.
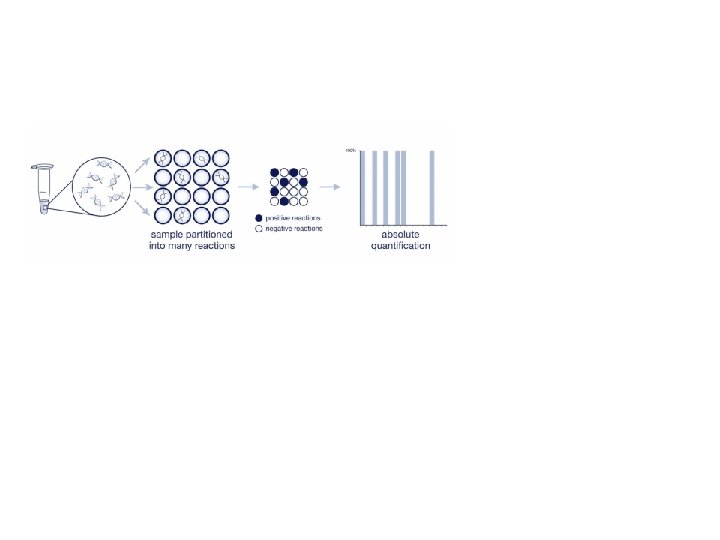
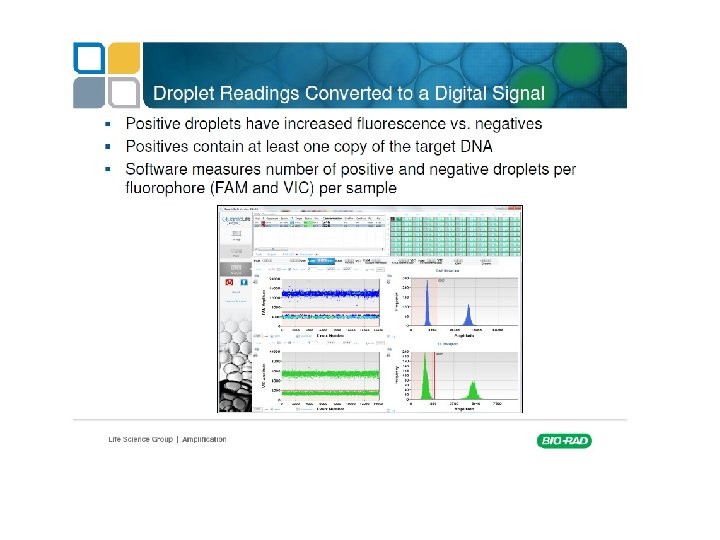
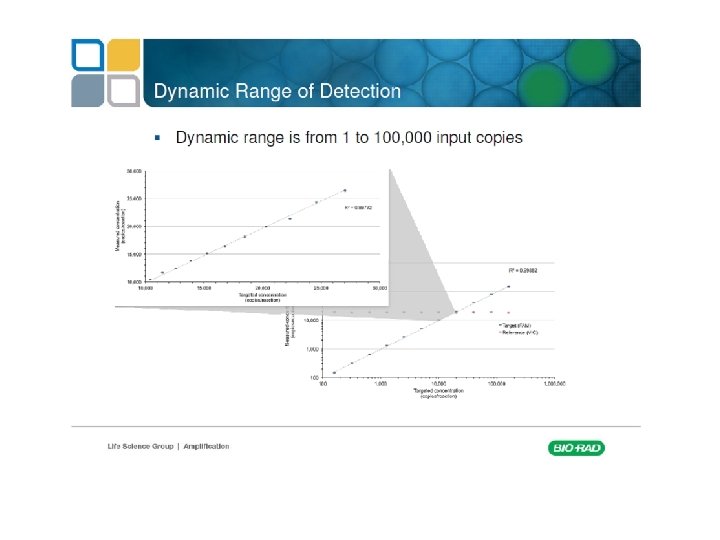
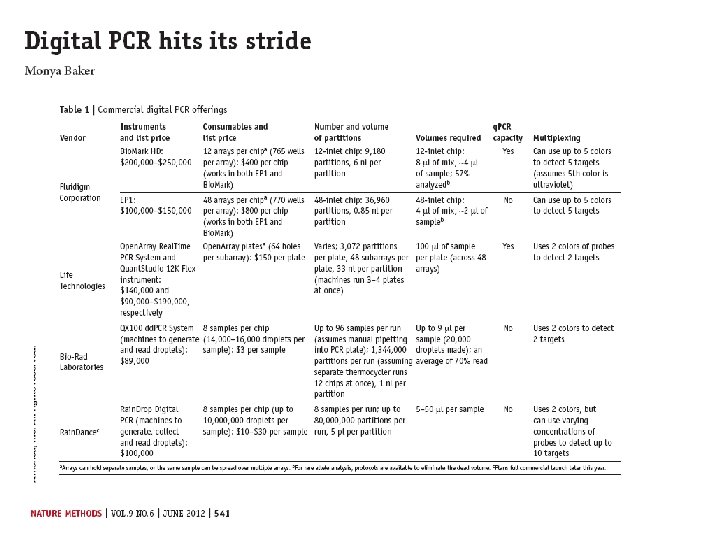
Digital PCR From Relative quantity to absolute quantity
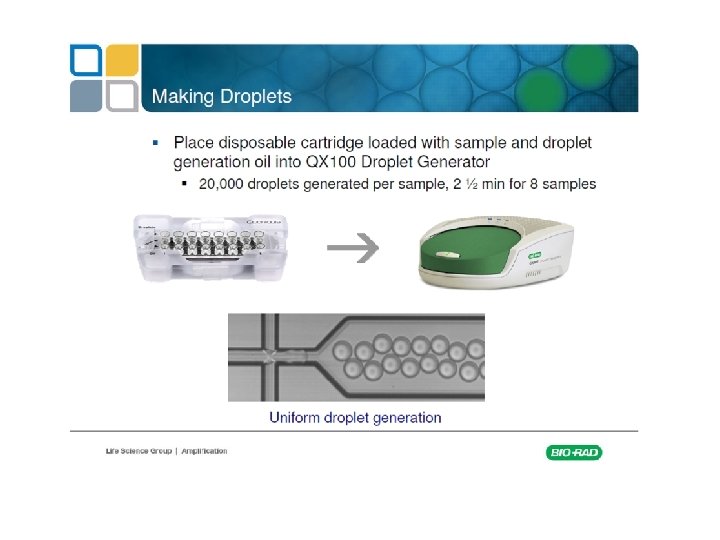
Commercially available machines Fluidigm Quanta. Soft (Life Technologies)
Rain Dance Bio Rad QX 100