Introduction to Gene Prediction Bioinformatics Lecture 10 Applications

Introduction to Gene Prediction Bioinformatics Lecture 10 Applications of Bioinformatic and Genomics/proteomics 2017

Let’s assume that you sequenced a large genomic segment. What to do next?
 atgcaggagattgagggactggcacagagatctccacaaaaccaggtaacatctgtgttccagggaacagaatagaaaa t ggacccagggctgtctctgacagggtgagcaactggagtgggaaagatctgggggacccaggagggtggagggact ggaactgggacctgaaggaggggctgcgcttcagcctcctcctcccactgcccctgcccccaggagcccttggtg aggaggaggccccagctctgtgatgcaggcctgggctccagtccctagtcccaaaagcgatgcccagggctcagttgctt gagggcaacattgcacttcagatggaaaaaatgctctttcctctgaagagccctagtgccacgtggctgggccccagctc cactccctggattgtggatttcatcctcagtgtggagcctgggcttcctattagccccctacctcccca")
Example of nucleotide sequence (from your assignment) atgcaggagattgagggactggcacagagatctccacaaaaccaggtaacatctgtgttccagggaacagaatagaaaa t ggacccagggctgtctctgacagggtgagcaactggagtgggaaagatctgggggacccaggagggtggagggact ggaactgggacctgaaggaggggctgcgcttcagcctcctcctcccactgcccctgcccccaggagcccttggtg aggaggaggccccagctctgtgatgcaggcctgggctccagtccctagtcccaaaagcgatgcccagggctcagttgctt gagggcaacattgcacttcagatggaaaaaatgctctttcctctgaagagccctagtgccacgtggctgggccccagctc cactccctggattgtggatttcatcctcagtgtggagcctgggcttcctattagccccctacctcccca gtgtcccaccctcacccccgcatgggaggaagaggagcagcaggaaggtaaggaaccctcag cccagccccacagagaaagattctctcttcttttcctcccctcatttccacttttcctaagtaatgagagactctcctgc gatgggaaatctcatcatccctctagggaagggcagagcaggggtgggagtgggcagaggattccatcctcagctc ccagactatctgtgacatgcgcgggaggcatcagggcaaaacccaacactggactcagtggcgaggaccagatcaggaga ggggtgaggtctctgtgggaggacaggccccagctctggctcatcagcccctttccaaggcaggtgcctgggggtccagg ctcctctgtgtggggtgatctgggagctgtgctgaacccctgagagcctcccgctagagcctggattcacgtctggcctc ctctgaagcagaatcctactggcagctcagaggtgcctgtaggcctgagcctgggtatgccttcagcggaggagaaggga ctggggatatccagggtggagtcacacagaaaacccctgcttgcatttccctaagaaggaaaacatttcatca atggtaaacatgagtaaaagaaacgcagagctccctgagctacaggcagaaagccgtgtccttcaagagcactgcg tgtgtgcagctggtctgggaagaggagactgagaactggtcccagcccctcacccttgcctctcggcatcaaagga agagataggggaggcccaggagcaggaagaagatcccagctctgaattgtgaggccctactaatccctgagactccccag aggtgactgagcatcgtctgtccctgagggaatcatttgcagaggcctgagagggaagctcctgggaaatcagaac ccggggcctcctcatattccctgtgggaatgaagccatgggcccgggaaccggtattgcccataagggggcgctgtgggg aagggaccaaggcctgccaggatatggggggaggtcagggagaccccaggtccacgtggacagctgttctcctcggccat ggctgactcttcttagagaccaccccatccactctccccacaagatggttgtgacacccatgaggggttgtacatggaag cactttttgcaaatgacaaagttctgcacaatgcacaccttgctgtcaccattggggccatgtggccttggacacagatg catgggcttcagggtctagttccccatggtgtcccctaaagagacatccaccactcagcctcccatgaggttcgctggca ccgccctccccgccataactctgtctccttacttccagcttgcagaatcctcctgagggagctggaggagactcaggacc tgaccaaccttctggaaagatgagattccccttctctccatgtcgtcctttctaccagggcctctgaggcagccccaggg tccagtgttgacctgggaggtcctgggaagaggcgggagtggactgaagccctgggcacagagctc tgtgaagccagcagtgggggagtggaggnnnnnnnnnnnnnnnnnnnnnnnnnnaaaccatcattgtcagcaaactatcgcaagga caaaaaaccaaataccacatattctcacacataggtgggaattgaacaataaggcattgtttctttatgaggaattagct

Mark Borodovski lab Gene. Mark http: //opal. biology. gatech. edu/Gene. Mark/ Cucumber genome 2009 Director, Center for Bioinformatics and Computational Genomics Georgia Tech, Atlanta, GA

Three steps for your task • Find a gene prediction program • Let your computer to work for you. • Take the results from the output file

How to find a FREE gene prediction program? • • Go to www. google. com Type “gene prediction program”, or “gene finding”, or “other_keywords” • D. Mount “Bioinformatics” pp. 339 -340 (>40 useful web sites)

Which program is the best? • Different programs give different results! Drosophila melanogaster – 14, 500 genes C. Elegans (nematode) - 18, 500 genes

INPUT: File with your sequence OUTPUT: File with predicted genes Parameters for the program which should be chosen by the user

What is important for gene prediction? Me 1. Understand the Genome Biology 2. Understand the computational algorithms Dr. Samuel Shepard Next two lectures

Three different types of genes and three different algorithms for their prediction 1. Protein coding genes 2. r. RNA and t. RNA genes 3. Other non-coding RNA genes (no sequence homology supercomputers are required)

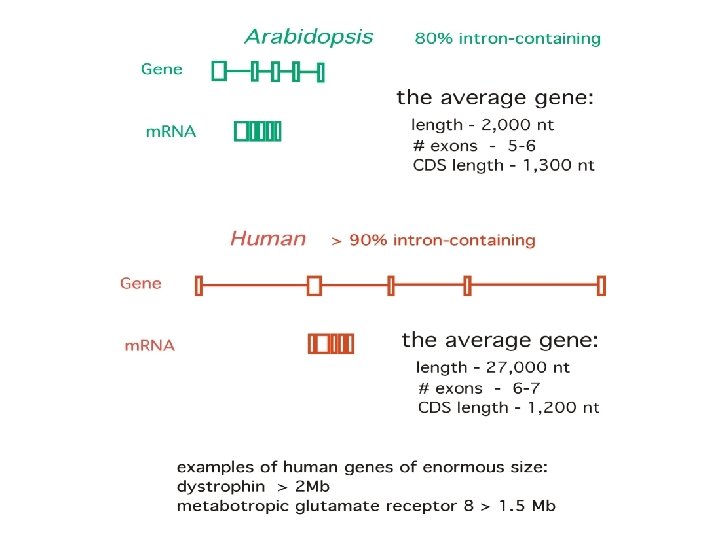
Gene finding algorithms for protein coding genes Two types of genome organization Prokaryotes Eukaryotes >90% of the genome are coding sequences Mammals: only 1% of their genome codes for proteins

Gene prediction in eukaryotes • Vast majority of the genomic DNA is noncoding sequences • Many genes have exon/intron structure

Genes have non-randomness in their nucleotide sequence patterns, which is the main clue for their prediction

Structure of m. RNAs Ribosome …ATG… 5`-UTR ATG…………. TAA CDS 60% GC 52. 5% GC Signals that regulate Translational control Coding information With 3 -nt periodicity AAAAA… 3`-UTR 44% GC Signals that regulate sub-cellular localization and stability

Gene structure GC-rich Intron AVERAGE Misleading due to CG-content = 42% Isochores !!!

sphingomyelin synthase 1 NM_147156 3`-UTR CDS Cow? Dog? Mouse? 5`-UTR exons
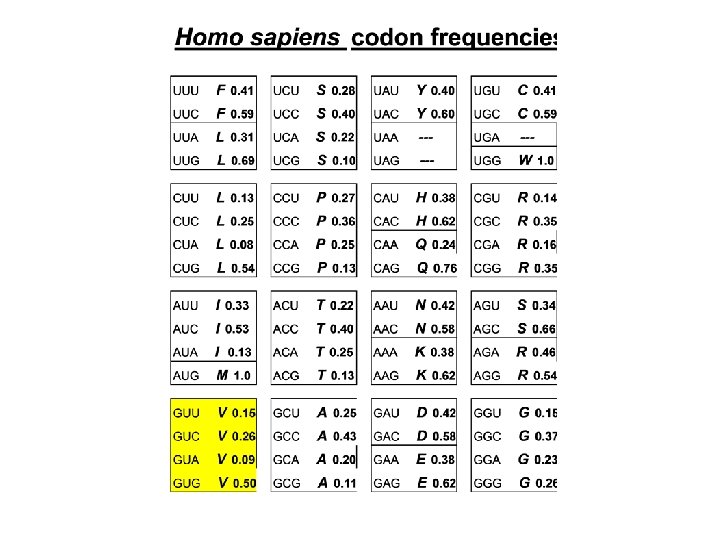
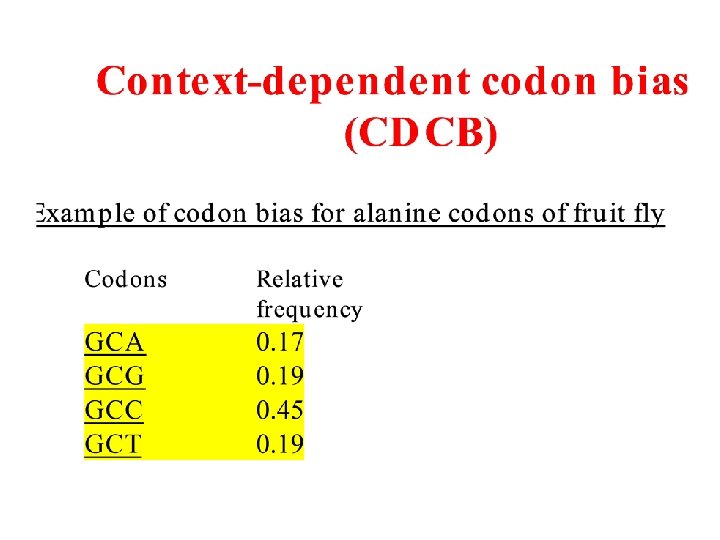
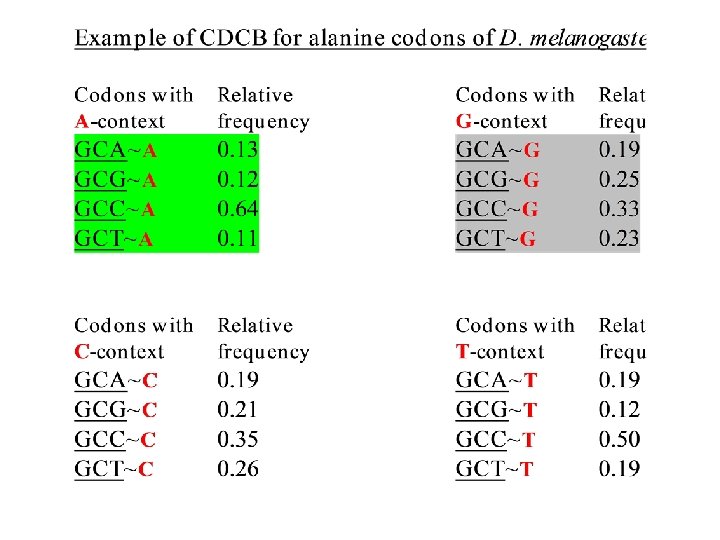
 ≠ F(x) × F(y)")
Amino acids also have preferable and nonpreferable neighbors F(xy) ≠ F(x) × F(y)
 set of exons and")
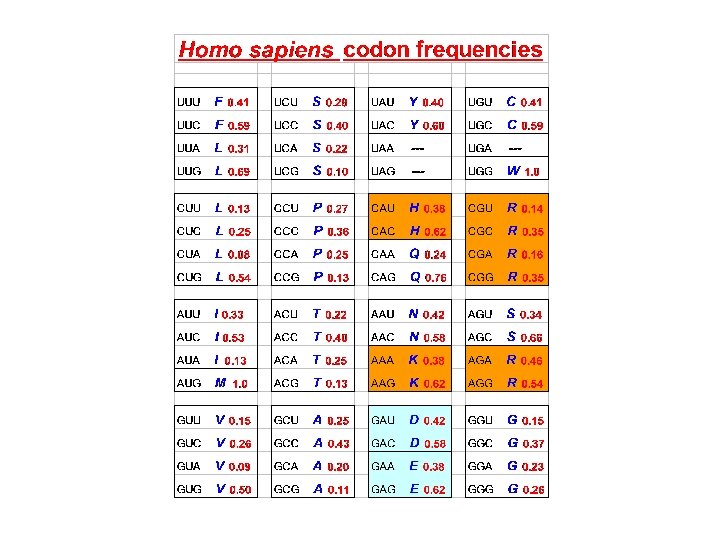
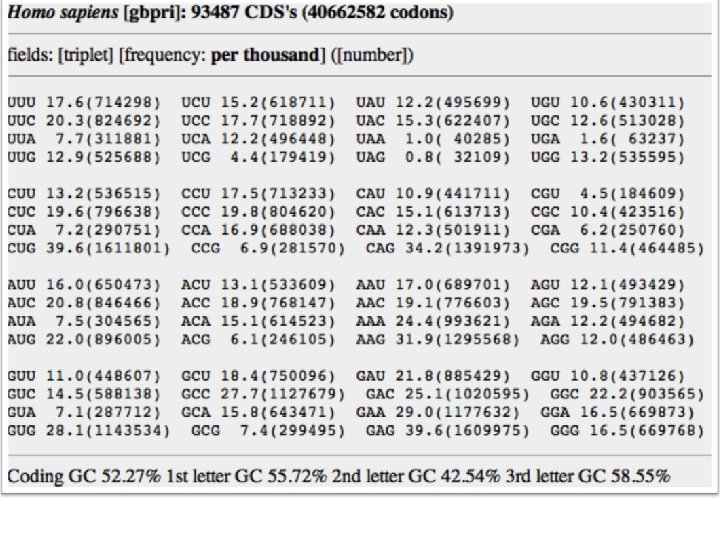
Training-based approach for discrimination of exons vs introns Initial (pre-known) set of exons and introns is required to calculate the discrimination rules (learning sample) coding potential, CP(xyz), for the triplet xyz, was calculated by one of the simplest forms using equation: CP(xyz) = log 10(Fc(xyz)/Fi(xyz)) where Fc(xyz) is the frequency of the triplet xyz inside coding exonic regions, and Fi(xyz) is the frequency of xyz inside introns. If CP(xyz) has a positive value, the xyz triplet is more abundant in exons versus introns. When CP(xyz) is negative, the opposite is true and xyz is more abundant in introns.

Coding potential for triplets in the human genome

How to calculate CP for a new test sequence Your input sequence ACTGGGCTAC…. . CP = CP(act) + CP(ctg) + CP(tgg) + CP(ggg) + +CP(ggc) + CP(gct) + CP(cta) + CP(tac) +… Finally, normalize by length: CP/L

Examination of the human test set of introns and exons usnig CP-values

How to get a training set of exons and introns? • Take it from the closest species which genes are well characterized. Yet some errors could be expected (fly and bee) • Get EST database for your species (additional sequencing of m. RNAs) • Get this training set from your genomic DNA by BLASTing it against evolutionary conserved protein sequences

Ab initio gene identification in metagenomic sequences • Sophisticated modern programs can perform “self-training” on sufficiently large genomes without the aid of pre-annotated training sequences. However, even such ab initio gene finders must still be instantiated with initial model parameters.

Recommended literature: DNA Composition, Codon Usage and Exon Prediction Chapter published in "Genetic Databases", M. J. Bishop ed. , Academic Press, 1999 Roderic Guigó Informàtica Mèdica, Institut Municipal d'Investigació Mèdica and Departament d'Estadística, Universitat de Barcelona February 7, 2000 http: //genome. imim. es/courses/Seq. Analysis/Gene. Identification/Search. Content/index. html

Coding potential Example of a coding sequence and 6 -mer oligonucleotides in two different reading frames (green and red) ATG TGC AAC GGG GCT AGG CAA GAT A T G C G T G C A A Due to codon bias and context dependent codon bias, distributions of oligonucleotides in different reading frames are distiguished from each other. Distribution of 6 -mer oligonucleotides in the real reading frame has unique features (coding potential).

Coding potential Graphical representation of the coding potential threshold G E N E 1 kb 2 kb Sequence

Average Mutual Information Ivo Grosse et al. 2000 exons introns


Human m. RNAs Any type of Restriction sites: AGTC GGCC ATGC TTCC ATGG etc

exons introns

Accuracy of exon/intron discrimination by 9 algorithms

Av. Mut. Info. pl -> Av. Mut. Info_pl. txt http: //bpg. utoledo. edu/~aprakash/Gen. Prediction_Php/Av. Mut. php Cu Ign rrentl ore y th this e pr ho ogra me wo m do rk ass es no ign t wo me nt rk du in t e t he o F vid irew eo all con flic ts! Do not change Play with this parameter

Prokaryotic genes • No introns inside protein coding genes! • The length of CDS might be small

Frequencies of stop codons in 6 possible reading frames 1 2 3 4 5 6 - Stop codon Probablilty of a stop codon at a particular triplet: P = 3/64 Probablilty that random sequence has an open reading frame coding n aa: P = (61/64)n n = 100, P = 0. 008 n = 200, P = 6. 7 * 10 -5 n = 300, P = 5. 5 * 10 -7 n = 400, P = 4. 5 * 10 -9 n = 500, P = 3. 7 * 10 -11

Important gene prediction procedures 1. Comparison of the genomic sequence with m. RNA and EST databases of the same species 2. Obtaining of all possible protein sequences by translation of the genomic sequence in 6 RF followed by comparison of these possible protein sequences with the entire protein database (Swiss. Prot)

Problems 1. About 20% of bacterial genes represent unknown proteins with no homology to any sequence from protein database. Many of these genes are short (~100 aa). 2. Many low expressing genes are absent in EST databases

Harrow J, Nagy A, Reymond A, Alioto T, Patthy L, Antonarakis SE, Guigó R. Identifying protein-coding genes in genomic sequences. Genome Biol. 2009 Jan 30; 10(1): 201 http: //genomebiology. com/2009/10/1/201
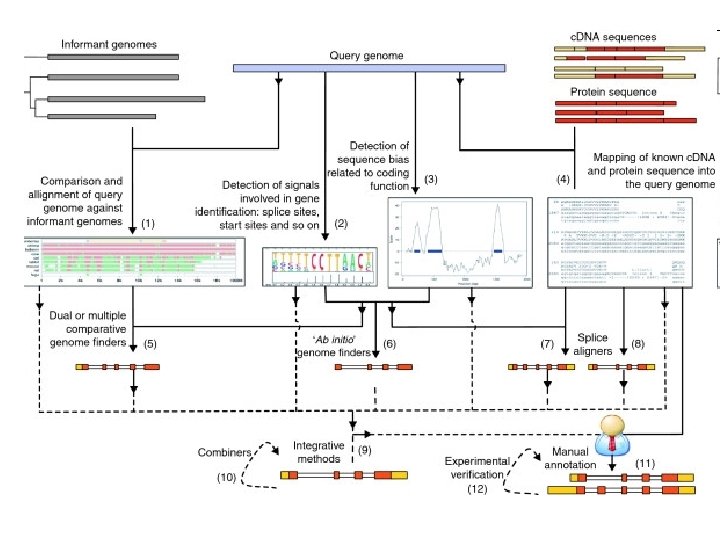

Assignment for L 10 ABPG 2017 Spend 2 hours reading about Markov Models in Guigó paper: “DNA Composition, Codon Usage and Exon Prediction” (excellent explanation). You will work with MM next two lectures! http: //genome. crg. es/courses/Seq. Analysis/Gene. Identification/Search. Content/index. html Or next link if first does not work (the paper is also available from the web/blackboard) http: //genome. crg. es/courses/Seq. Analysis/Gene. Identification/Search. Content/
- Slides: 45