Introduction THE HAEMOGLOBIN MOLECULE Human haemoglobin is formed


Introduction THE HAEMOGLOBIN MOLECULE Human haemoglobin is formed from two pairs of globin chains each with a haem group attached.



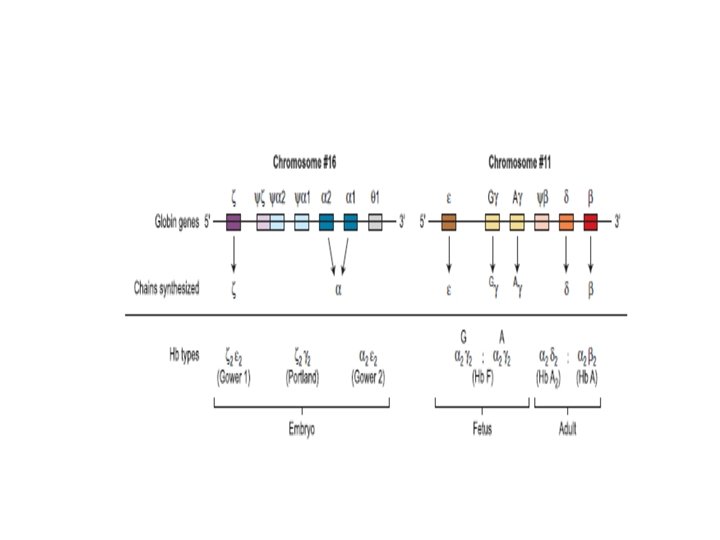
Seven different globin chains are synthesised in normal subjects: two, ε and ζ, are characteristic of the embryo and contribute to four transient embryonic haemoglobins referred to as haemoglobins Gower 1, Gower 2, Portland 1 and Portland 2.

The individual chains synthesised in postnatal life are designated α, β, γ and δ.

Haemoglobin F is the predominant haemoglobin of fetal life and comprises the major proportion of haemoglobin found at birth. Haemoglobin F : (α 2γ 2) •

Haemoglobin A is the major haemoglobin found in adults and children. Haemoglobin A : (α β ) 2 2 Haemoglobin A 2 has two α chains and two δ chains (α δ ). 2 2

Haemoglobins A 2 and F are found in small quantities in adult life (approximately 2– 3. 3% and 0. 2– 1. 0%, respectively). The adult proportions of haemoglobins A, A 2 and F are usually attained by 6– 12 months of age.

The α chain is thus common to all three types of haemoglobin molecule. -α chain synthesis is directed by two α genes, α 2 and α 1, on chromosome 16. -β and δ chain synthesis by single β and δ genes on chromosome 11. γ chain synthesis is directed by two genes, Gγ and Aγ, also on chromosome 11.

Inherited disorders of haemoglobin are the commonest single gene disorders, with an estimated carrier rate of 7% among the world population.

Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the two. .

-Quantitative defects cause thalassaemia syndromes, -Qualitative changes, referred to as haemoglobin variants, cause wide range of problems, including sickle cell disease , unstable haemoglobins, decreased oxygen affinity, increased oxygen affinity, and methaemoglobinaemia.

the majority of qualitative mutations cause • no significant change in haemoglobin properties or clinical problems.

Collectively, the clinical syndromes resulting from disorders of haemoglobin synthesis are referred to as ‘Haemoglobinopathies’. They can be grouped into three main categories: 1. Those resulting from a genetically determined structural variant of haemoglobin , such as haemoglobin S. 2. Those owing to failure to synthesise one or more of the globin chains of haemoglobin at a normal rate, as in the thalassaemias. .

3. Those owing to failure to complete the normal neonatal switch from fetal haemoglobin (haemoglobin F) to adult haemoglobin (haemoglobin A). **Hereditary persistence of fetal haemoglobin (HPFH)

An individual can also have a combination of two or more of these abnormalities (e. g. a variant haemoglobin can be synthesised at a reduced rate). e. g. Hb E

The clinical syndromes produced by haemoglobin abnormalities: clinical syndromes produced by Syndrome Abnormality haemoglobin abnormalities Haemolysis Crystalline haemoglobins (Hb S, haemoglobin abnormalities C, D, E, etc. ) Unstable haemoglobin Thalassaemia α or β resulting from reduced globin chain synthesis Familial polycythaemia Altered oxygen affinity Methaemoglobinaemia Failure of reduction (Hb Ms)

Thalassaemias.

The thalassaemia syndromes are a heterogeneous group of inherited conditions characterised by defects in the synthesis of one or more of the globin chains that form the haemoglobin tetramer. 7

They are encountered in every population in the world but , β‐Thalassaemia is more common in the Mediterranean region while α‐thalassaemia is more common in the Far East.

The clinical syndromes associated with thalassaemia arise from the combined consequences of inadequate haemoglobin production and of unbalanced accumulation of one type of globin chain. The former causes anaemia with hypochromia and microcytosis; the latter leads to ineffective erythropoiesis and haemolysis.

Clinicopathological manifestations range from completely asymptomatic microcytosis to profound anaemia that is incompatible with life and can cause death in utero

Clinically the main syndromes are: -Thalassaemia major: Transfusion dependent -Thalassaemia intermedia : Non‐transfusiondependent thalassaemia with a moderate degree of anaemia due to a variety of genetic defects -Thalassaemia minor, usually due to a carrier state for α‐ or β‐thalassaemia.

This clinical heterogeneity arises as a result of the variable severity of the primary genetic defect in haemoglobin synthesis and the coinheritance of moderating factors, such as the capacity to synthesise increased amounts of haemoglobin F.

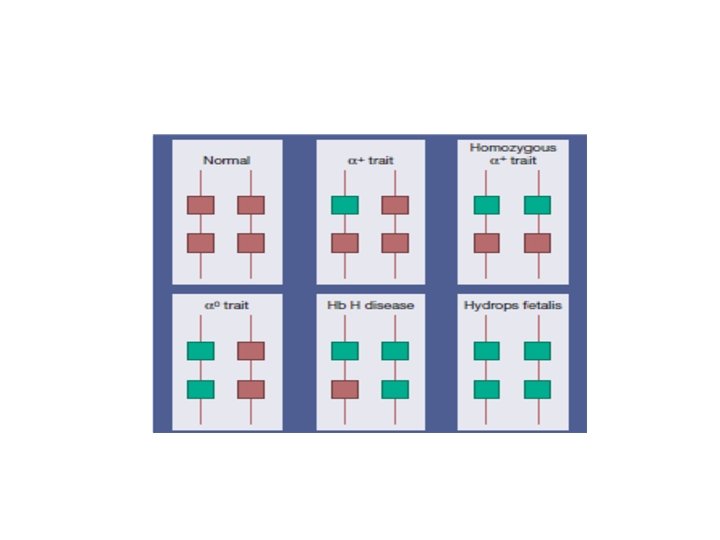
α thalassaemia syndromes These syndromes are usually a result of deletions of one or more α genes, although some mutations described are nondeletional. The clinical severity is related to the number of the four α‐globin genes missing or inactive.

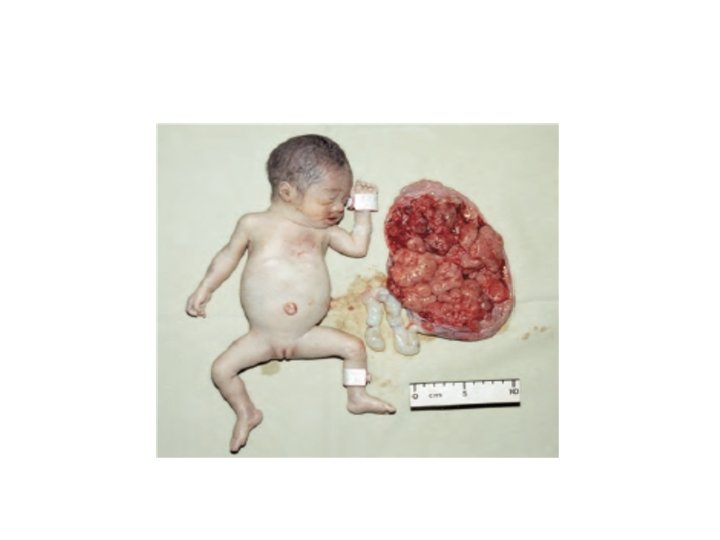
Haemoglobin Bart’s hydrops fetalis Loss of all four genes completely suppresses α‐chain synthesis and because the α chain is essential in fetal as well as in adult haemoglobin this is incompatible with life and leads to death in utero -occurs mainly in people from Southeast Asia but is also -occasionally observed in people from Greece, Turkey and Cyprus. -.

An affected fetus will be stillborn or will die shortly after birth. Sever anaemia and oedema are the hallmarks of this condition. Women carrying a hydropic fetus have a high incidence of complications of pregnancy. Prenatal diagnosis should be offered for women at risk of having a fetus with haemoglobin Bart’s hydrops fetalis
")
Hb H disease -Three α gene deletion A moderately severe (haemoglobin 70– 110 g/L) microcytic, hypochromic anaemia with splenomegaly. -haemoglobin H (β 4) Haemoglobin H can be detected in red cells of these patients by : 1 -electrophoresis 2 -reticulocyte preparations


α thalassaemia trait 0 * a chromosome has no functional α genes * − −/αα. *microcytosis. *The Hb may be normal or slightly reduced.

α+ thalassaemia trait *where one of the two globin genes on a single chromosome is absent or fails to function. * α+ thalassaemia heterozygosity −α/αα *α+ thalassaemia homozygosity −α/−α

α+ thalassaemia trait can be completely silent, or there may be borderline microcytosis with a slightly reduced or normal MCH

Haematologically, homozygosity for α+ thalassaemia trait resembles heterozygosity for α 0 thalassaemia trait, but the genetic implications are very different.

Both α+ thalassaemia trait and α thalassaemia trait are more difficult to diagnose than β thalassaemia trait because there is no characteristic elevation in haemoglobin A , and haemoglobin H inclusions may not be demonstrated. 0 2

Definitive diagnosis of α thalassaemia trait is more reliably made with the use of DNA techniques or globin chain biosynthesis studies.

-Uncommon non‐deletionalforms of α‐thalassaemia e. g. Hb constant spring.

THANK YOU

- Slides: 43