Interstitial lung Diseases By Dr Azza Farag El


Interstitial lung Diseases By Dr. Azza Farag El- Toney Professor of Chest Diseases
 l")
Items of presentation l Anatomy of interstitium l Definition of interstitial lung disease(ILD) l Pathogenesis of ILD l Classification of ILD l Diagnosis of ILD l Treatment of ILD

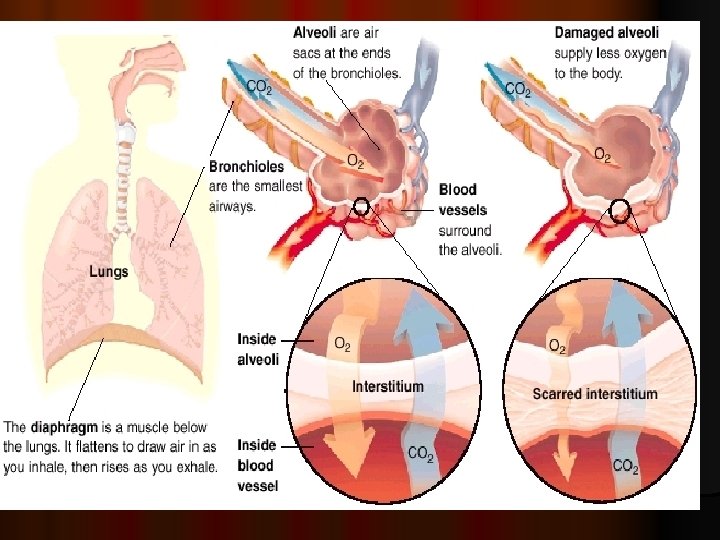
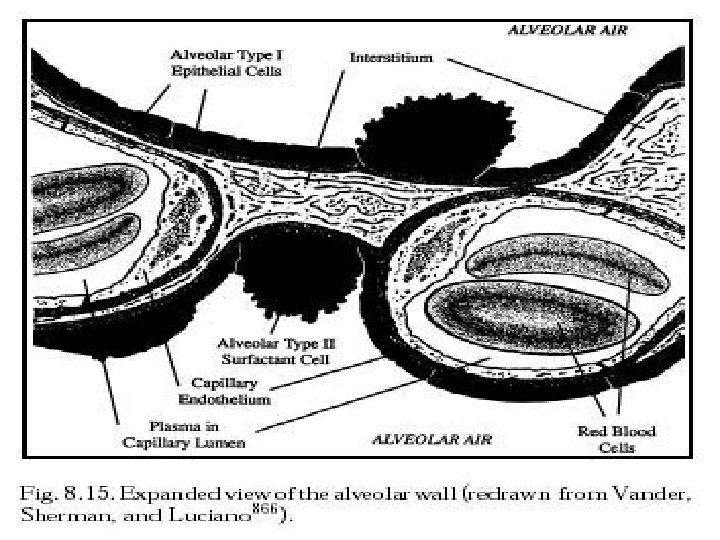
Interstitium: is defined as continuum of loose connective tissue throughout the lung. It is the supporting framework of the lung. It contains elastin , fibroblast and tissue macrophages may be found in interstitial space.
 the bronchovascular (axial), surrounding the bronchi,")
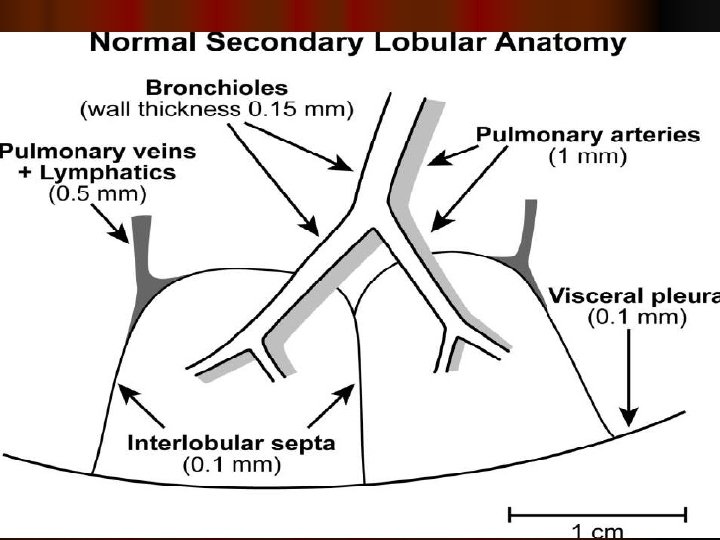
l It composed of three subdivisions: l (i) the bronchovascular (axial), surrounding the bronchi, arteries, and veins from the lung hila to the level of the respiratory bronchiole l (ii) peripheral , the subpleural, situated beneath the pleura, as well as in the interlobular septae l (iii) the parenchymal (acinar), situated between the alveolar and capillary basement membranes (intralobular interstitium).
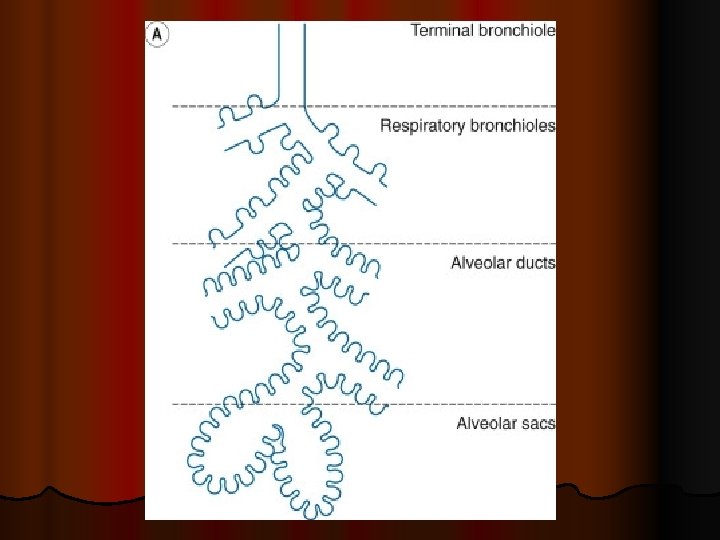

Secondary pulmonary lobule

 comprise a heterogeneous group")
Interstitial lung disease: ILD or diffuse parenchymal lung diseases (DPLDs) comprise a heterogeneous group of lung disorders characterized by various degrees of inflammation and pulmonary fibrosis occurring predominantly in the interstices or supporting structures of the lung.

l The term interstitial is misleading since most of these disorders are also associated with extensive alterations of alveolar, pulmoanry interstitium and airway architecture.

l ILDs are a group of diseases with similar clinical, radiologic, and lung function presentations. l Characterized by: l 1. Dyspnea with exertion then at rest l 2. Radiologically diffuse bilateral infiltrates. l 3. Histologically by distortion of the gas exchanging units. l 4. Physiologically by restrictive pulmonary function and impaired oxygenation

l Pathogenesis of ILD l ILD is a multifactorial disorder and its disease susceptibility is associated with genetic and environmental factors such as smoking, micro-aspiration, inhalation and the use of some medications. l Different susceptibility genes are found to be associated with each subset of ILD, suggesting the presence of heterogeneity in ILD.

Pathogenesis of ILD
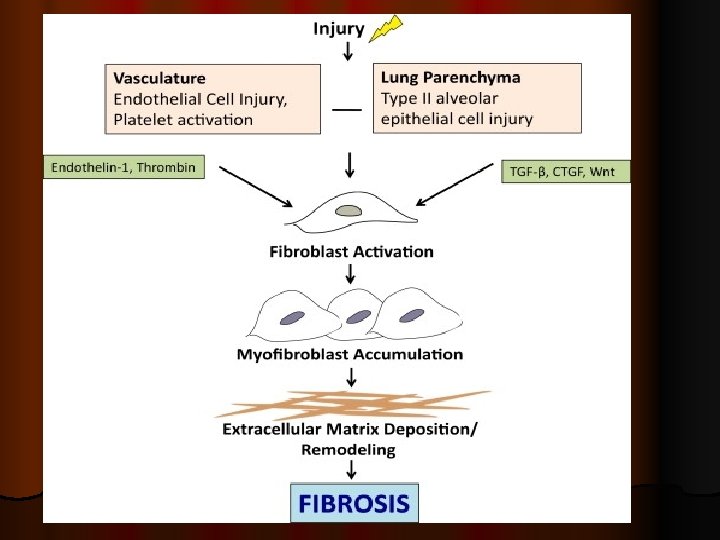

Pathogenesis of ILD Exposure to causal agents induce immune inflammatory response which lead to accumulation of inflammatory cells within the alveolar structures. These inflammatory cells produce different mediators leading to accumulation of increased numbers of neutrophils and eosinophils which release reactive oxidant species and proteases which produce local lung damage or injury.

As the disease progress, there is loss of type I alveolar cells and capillary endothelial cells with proliferation of type II cells and interstitial fibroblast with accumulation of type I collagen, and the transformation of fibroblasts into myofibroblasts. Fibroblasts and myofibroblasts are key effector cells in fibrogenesis, and myofibroblasts secrete extracellular matrix proteins fibronectin fibrosis.

Classifications of ILD l Recently, in 2013 , a multidisciplinary panel of American Thoracic Society/European Respiratory Society (ATS/ERS) members published a revised classification of ILDs based on their clinical, radiologic, and histopathologic findings. l Travis WD et al. Am J Respir Crit Care Med. 2013; 188: 733– 48.
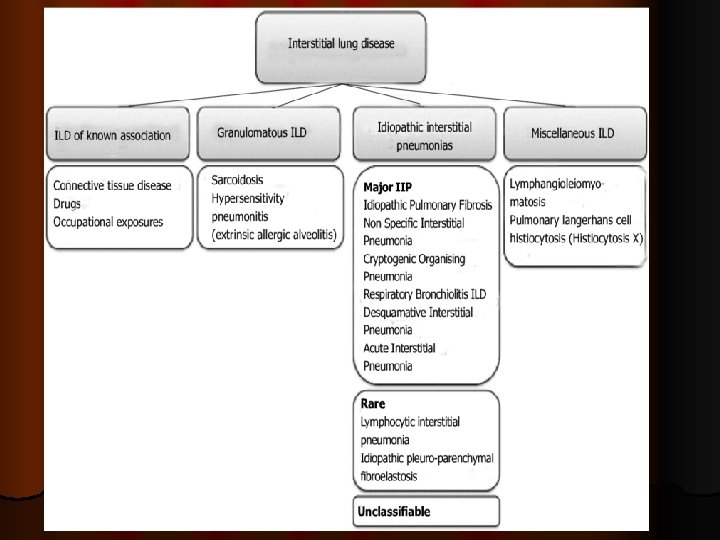

l I-ILDs of known cause: -Environmental exposure like occupational lung diseases ( coal worker’s pneumoconiosis, silicosis, asbestosis). -Radiation exposure to the chest
. -")
- Drugs( cytotoxics as busulfan, bleomycin , methotrexate, nitrofurantoin, sulfasalazine, gold, penicillamine, amiodarone). - Connective tissue disease related (rheumatoid, systemic sclerosis, SLE).
 1 - Major IIP (Idiopathic pulmonary fibrosis(IPF), idiopathic non")
l II- Idiopathic interstitial pneumonias(IIPs) 1 - Major IIP (Idiopathic pulmonary fibrosis(IPF), idiopathic non specific interstitial pneumonia (NSIP). - Smoking related IIP (desquamative interstitial pneumonia (DIP), respiratory bronchiolitis interstitial lung disease (RB-ILD). -Acute IIP ( acute interstitial pneumonia (AIP) , cryptogenic organising pneumonia(COP). 2 -Rare IIP ( lymphocytic interstitial pneumonia (LIP), idiopathic pleuroparenchymal fibroelastosis(PPFE). 3 - Unclassifiable

l III- Granulomatous lung disorders l Sarcoidosisis a systemic disorder of unknown cause characterised histologically by the presence of noncaseating granulomata in affected organs. Thoracic manifestations, the most common cause of morbidity and mortality, occur in 90% of the patients and 20% of them develop chronic fibrotic lung diseases. -Hypersensitivity pneumonitis or extrinsic allergic alveolitis (inhalation of bacterial or fungal spores contaminate grains or hay (farmer's lung), or inhalation of antigenic material in bird’s feathers or excreta ( pigeon-breeder's lung).
 -Pulmonary langerhan cell’s histocytosis (PLCH).")
IV- Other forms of ILD: -lymphangioleiomyomatosis( LAM) -Pulmonary langerhan cell’s histocytosis (PLCH).

Epidemiology Rates of interstitial lung disease are somewhat higher in men than in women (at a ratio of 1. 5: 1). l The pneumoconioses (eg, silicosis) are much more common in men than in women, probably because of higher rates of occupational exposure l While some aetiologies of interstitial lung diseases women are much more likely to develop rheumatologic/connective-tissue disease than men and thus are more likely to experience pulmonary manifestations of those diseases also, LAM exclusively affect women. l

Some diseases are insidious in onset and gradual but unrelenting in progression (eg, similar to IPF), while other diseases are acute in onset (AIP, COP). l As regard age, most patients with IPF present in the fifth or greater decade of life. l Others forms of interstitial lung disease, such as sarcoidosis, LAM, connective-tissue disease– associated lung disease, primarily present in younger adults. l

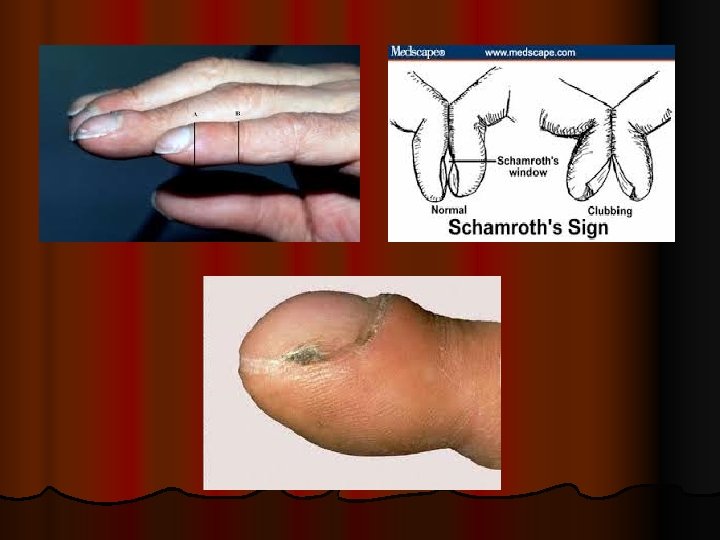
Symptom and Signs - - - Early: dyspnea on exertion, late: dyspnea at rest. In idiopathic pulmonary fibrosis (IPF), dyspnea begins insidiously and is often progressive. Cough, chest pain can occur ( it is due to pleurisy in CVD or pneumothorax complicating LAM and IPF ). Systemic symptoms include weight loss, lowgrade fevers, fatigue, arthralgias, or myalgias in sarcoidosis, CVD. . Digital clubbing is common with some diagnoses (eg, IPF and asbestosis) and may first be noted by the patient , however, it may also herald an underlying bronchogenic carcinoma.
")
chest exam may be unremarkable or Fine end-inspiratory pulmonary rales or crepitations (Velcro rales) are a common finding and may be difficult to distinguish from those auscultated in patients with congestive heart failure. - Wheezes may be heard and reflect airway involvement, as in sarcoidosis or in those with hypersensitivity pneumonitis. -

l Central cyanosis may be present if significant hypoxemia

l Disease-specific findings include the following: l Generalized lymphadenopathy often occurs with sarcoidosis, . l Cutaneous and articular findings are associated with rheumatologic disease.

Investigations 1 - CXR: Early it may normal, then reticular, nodular or reticulonodular shadows. Late ( end- stage), coarsely reticular, cystic and honeycomb patterns. l Sarcoid-associated interstitial disease often demonstrates symmetric hilar adenopathy. l IPF, asbestosis, and connective-tissue disease–related changes are most often basilar and peripheral in distribution.

l upper lung zone infiltrate predominance, as may sarcoidosis, chronic HSP, pneumoconioses, and drug-related DPLD due to gold or nitrofurantoin therapy.

Normal CXR

Bilateral reticulation

Bilateral nodular shadows

Extensive fibrosis with decrease lung volumes
 of the chest: CXR may be")
2 - High Resolution Computed Tomography ( HRCT) of the chest: CXR may be normal in up to 10% of patients with biopsy – proven interstitial disease where HRCT may be abnormal. High-resolution chest CT scanning is more sensitive than chest radiography It help in establishing diagnosis, in selecting type of biopsy and the optimal biopsy site.

reticular pattern are the most common findings On HRCT images , in idiopathic pulmonary fibrosis it is characterized by patchy, predominantly peripheral, predominantly subpleural, and bibasilar reticular opacities l A ground-glass pattern , It is an opacification that does not obscure underlying lung markings and is thought to be a favorable prognostic finding. l Honeycombing pattern is indicative of end-stage disease and carries a poor prognosis. l Nodular pattern suggests HSP, granulomatous disease (eg, sarcoidosis), l Cystic pattern may be seen in LAM , PLCH or histocytosis X. l

Normal lung window

Bilateral subpleural reticulation

Bilateral extensive ground glass opacity

Red circle points to honeycomb

Nodular pattern

Cystic pattern
: Restrictive ventilatory defect( vital capacity, FEV 1 (Forced Expiratory")
3 -Pulmonary Function Tests( PFT): Restrictive ventilatory defect( vital capacity, FEV 1 (Forced Expiratory volume in 1 st second) , FVC (Forced Vital Capacity) , normal Forced Expiratory volume in 1 st second / Forced Vital Capacity FEV 1/ FVC or even increase in late stages (> 70% predicted). TLC (total lung capacity), RV (residual volume). Decrease of diffusion capacity of the lung to carbon monoxide( DLCO), lung compliance.


4 - Blood studies: -Nonspecific: ESR, CBC may show mild anemia, raised CRP l angiotensin converting enzyme( ACE) is elevated in sarcoidosis. - RF, ANA , Ads. DNA more helpful with collagen vascular causes of interstitial disease. - Serum precipitins to organic dusts helpful in hypersensitivity pneumonitis.
 analysis often reveals hypoxemia (reduced partial pressure of")
5 - Arterial blood gas (ABG) analysis often reveals hypoxemia (reduced partial pressure of oxygen (Pa. O 2) < 80 mm. Hg ( normal 80 -100 mm. Hg) or respiratory failure = (Pa. O 2) < 60 mm. Hg on breathing room air. l Oxygen desaturation is common. l All of these findings may be worsened with exercise.
 : A way of sampling the epithelial lining")
6 - Bronchoalveolar Lavage ( BAL) : A way of sampling the epithelial lining fluid of the lower respiratory tract and analyzing the effector cells making up the alveolitis. .

7 - Lung Biopsy: Transbronchial and endobronchial lung biopsies may be diagnostic, particularly for sarcoidosis but frequently are not useful for other diagnoses. This is due to the patchy distribution of the majority of these diseases and so this type of biopsy may not be representative of the extent or intensity of the disease.

Thus surgical lung biopsy by open thoracotomy or video- assisted thoracoscopy are the methods of choice.

Treatment of ILD I-Pharmacologic agents that are prescribed to prevent progression and/or induce remission (if the specific disorder can respond to such)include Immunosuppressive anti-inflammatory agents are used to treat various forms of ILD.

l such as corticosteroids. This is particularly the case for some disorders such as cryptogenic organizing pneumonia (COP), , sarcoidosis, or cellular non-specific interstitial pneumonia (NSIP).

l When extensive fibrosis is present, such therapies may be less efficacious, especially for patients with IPF, for whom currently available immunosuppressive or antifibrotic therapies are not recommended

l some forms of CTD-associated ILD have been reported to respond to mycophenolate therapy, which also allowed a significant lowering of corticosteroid dosing.

l Treatment of IPF l The prognosis of IPF is generally poor, and the majority of patients have progressive loss of lung function and may suffer acute exacerbations with acceleration of lung function loss that often leads to death

Until recently, there were no specific treatment options for IPF other than lung transplantation. l The previous widespread practice of using immunosuppressive therapy (steroid, azathioprine)in IPF was found to be ineffective and associated with worse outcomes. l In 2014, FDA approved two antifibrotic agents (nintedanib and pirfenidone) were found to reduce the decline in lung function over 12 months. l

l II-Lung transplantation l Lung transplantation is an accepted form of treatment for patients with ILD that is progressive, clearly leading to respiratory failure, and refractory to otherapies. l Lung transplantation is the only form of therapy that may improve quality of life and survival for patients with IPF.

l III-supportive therapies (e. g. supplemental oxygen if indicated, (Pa. O 2 ≤ 55 mm. Hg or oxygen saturation as measured using pulse oximetry [Sp. O 2] ≤ 88%) at rest or with exercise. patients with pulmonary hypertension, Cor- pulmonale) l Pulmonary rehabilitation. l measures to relieve symptoms (e. g. cough, anxiety, depression, dyspnea) and l treatment of co-morbid conditions (e. g. anemia, sleep disordered breathing, GERD, infectious complications).

l Vaccination against influenza and pneumococcal infection should be encouraged in all patients with idiopathic pulmonary fibrosis.

MCQ l In patients with suspected idiopathic pulmonary fibrosis, the most valuable measure is: l A. Bronchoscopy B. Sedimentation rate C. Trial of steroids D. Video-assisted thorascopic surgery (VATS)

l all the following are features of interstitial lung disease except ; a. exertional dyspnea b. early productive cough c. digital clubbing d. coarse crepitations during inspiration

l Investigation of choice for detection and characterization of ILD is ? A- MRI B-CXR C-HRCT D- Ventilation perfusion scan

l A 40 year old woman is seen in the office complaining of shortness of breath in climbing a flight of stairs, which has been progressive over the past four months. Her physical examination is unremarkable. Her chest X-rays show bilateral hilar adenopathy, with a mild diffuse infiltrate in the lung fields. What is the most likely diagnosis: A. Idiopathic pulmonary fibrosis (IPF) B. Sarcoidosis C. Wegener’s granulomatosis D. Asbestosis

In interstitial lung diseases, lung function tests most often show: A. Reduced carbon monoxide diffusing capacity (DLCO) B. Increased total lung capacity (TLC) C. Airflow obstruction D. Elevated arterial PCO 2 l

l interstitial lung disease is associated with the following except a. sarcoidosis b. asbestosis c. carcinoid lung d. radiation exposure

l -Pulmonary function abnormalities in ILD include all of the following except? A- Reduced VC B- Reduced FEV 1/FVC C- Reduced diffusion capacity D- Reduced total lung capacity

- Slides: 72