INHERITED RENAL DISORDERS AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE






 polycystic kidney disease • Infants, adolescents, young adults • 1 in")






















")



- Slides: 45
INHERITED RENAL DISORDERS
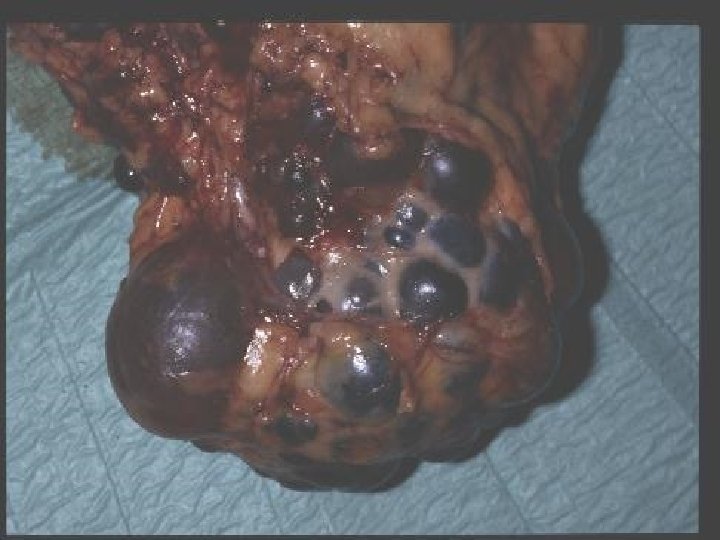
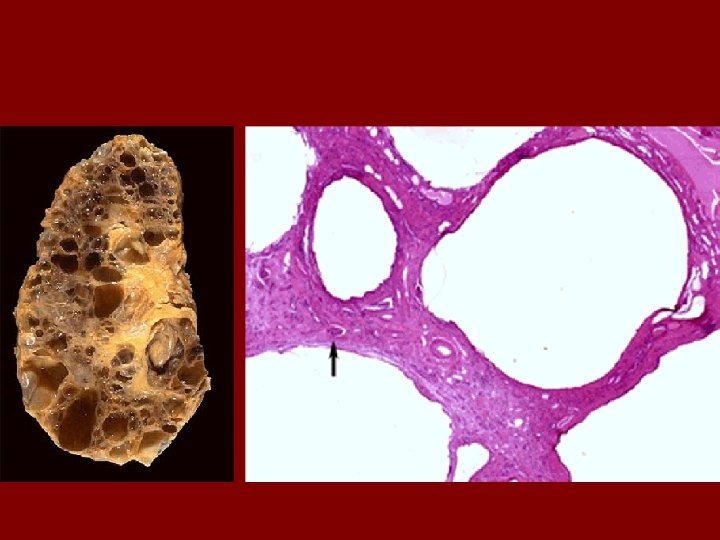
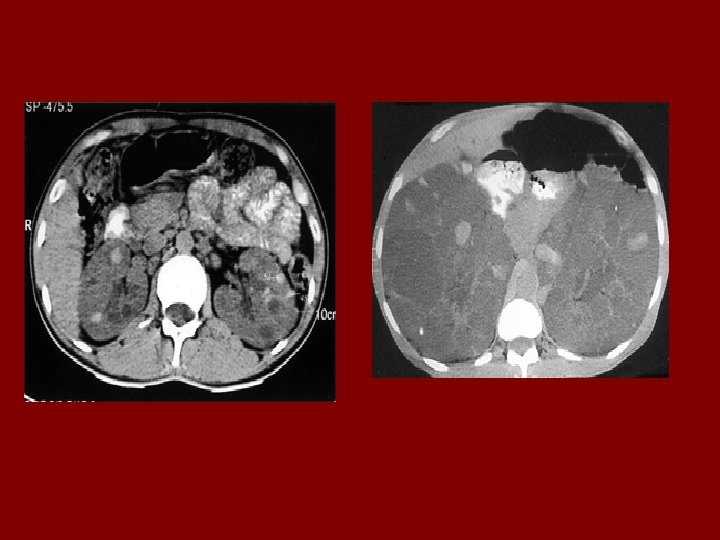
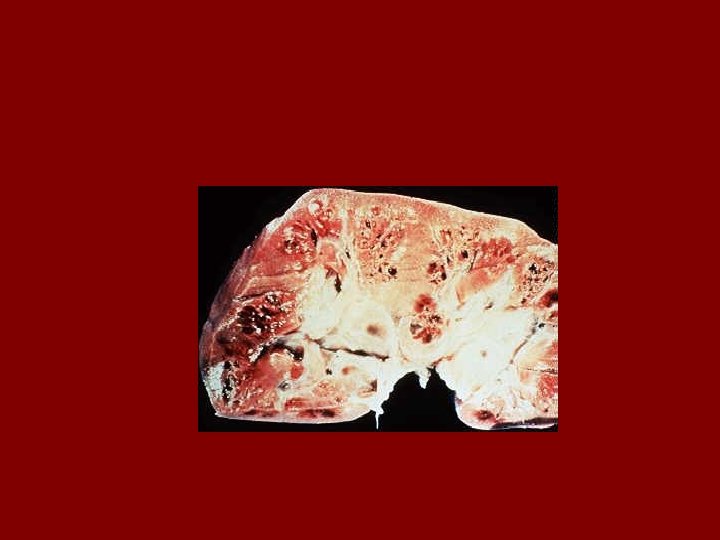
AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE • Prevalence: 1: 300 to 1: 1000 • 90% of cases are inherited, 10% are sporadic • Only 1 to 5% nephrons developed cysts • Cysts are in medulla and cortex • ADPKD causes symptoms in third or fourth decade • 50% of patients developing ESRD by age 60
RENAL Symptoms of ADPKD • Chronic flank pain • Acute pain indicates: infection (pyelonephritis- pyocyst) urinary tract obstruction sudden hemorrhage into cysts • Hematuria • Impaired renal concentrating ability • Nephrolitiasis in 15%to 20% • Hypertension in 75% adults
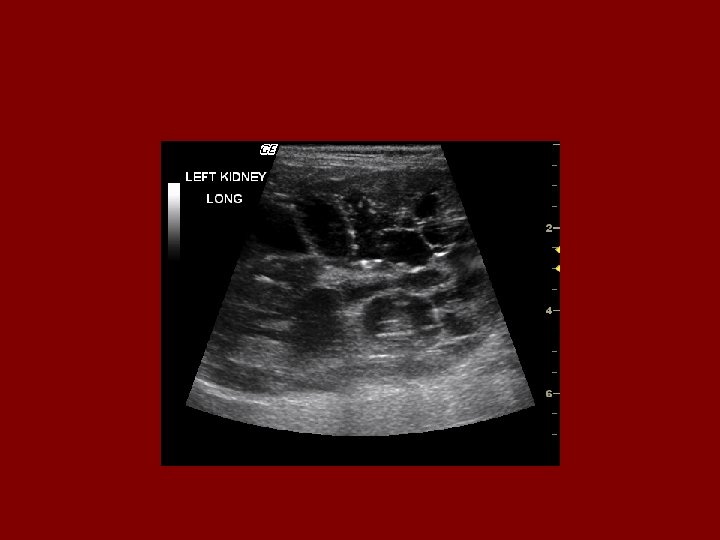
Diagnostic Criteria- Ultrasound • Age 15 -29 ………. . 2 cysts in one or both kidneys • Age 30 to 59 ……. 2 cysts in each kidney • Age >60 …………. 4 cysts in each kidney
EXTRARENAL SYMPTOMS • CYSTS in: Liver Spleen Pancreas Ovaries • Intracranial aneurism • Colonic diverticular disease • Mitral valve prolapse
Factors affecting progression to End Stage Renal Failure • • Hypertension Recurrent Haematuria UTI (in men) Massive Liver cystic Disease (mostly women) • More than 3 pregnancies • Age at symptomatic diagnosis • Sickle Cell Trait
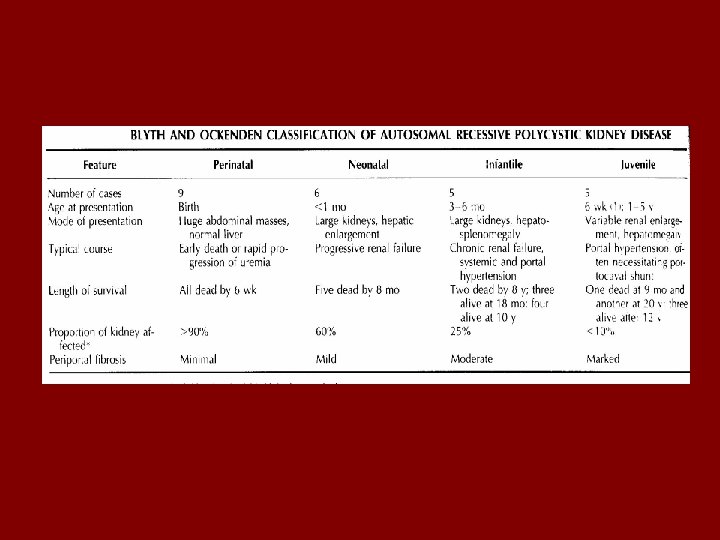
Autosomal recessive (infantile) polycystic kidney disease • Infants, adolescents, young adults • 1 in 20, 000 affected • 1 in 10, 000 in Finland • Up to 50% livebirths die within hours of birth • Those that survive neonatal period – 50% alive at 10 yo
• Most severe forms early in life • Less severe forms present later – Always bilateral – Always congenital hepatic fibrosis • AR – 1 in 4 chance – neither parent shows signs • Ch. 6 identified in all forms
Clinical features • Renal and liver pathology inversely related • Oligohydramnios • Potter’s facies • Respiratory distress –pulmonary hypoplasia
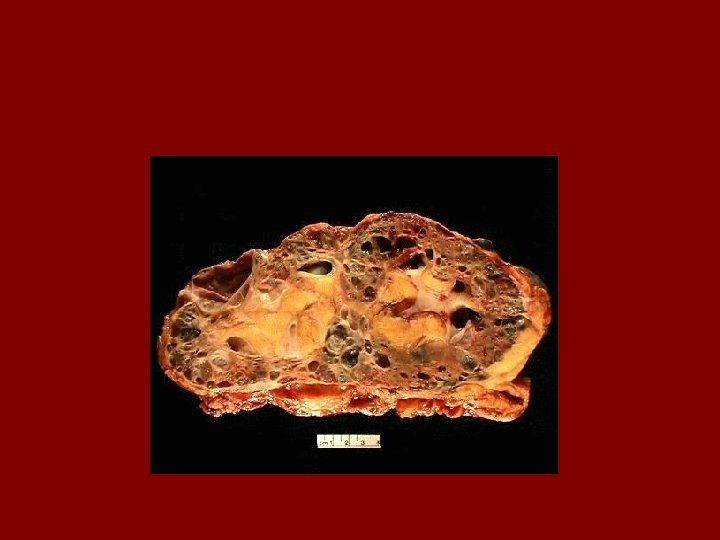
Histopathology • Retain fetal lobulation • Subcapsular cysts • Radial cortical cysts – dilated tubules
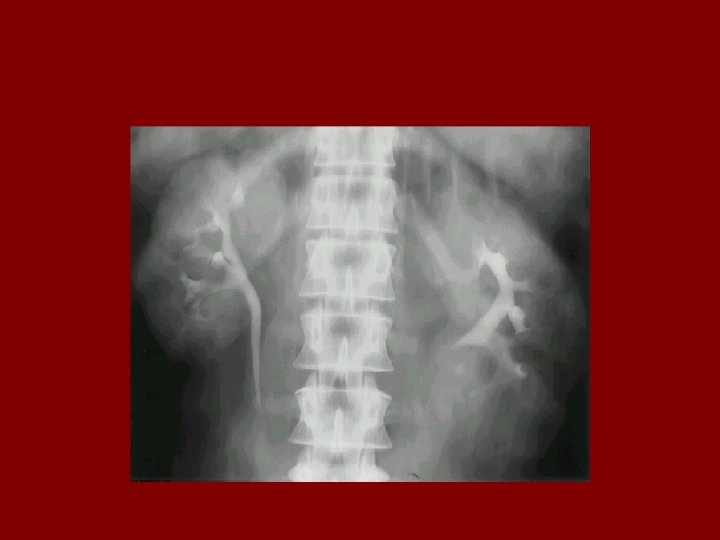
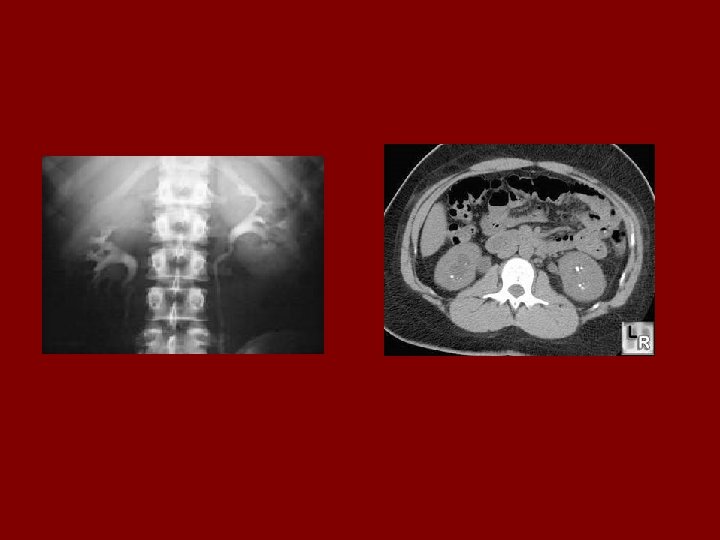
Evaluation • In utero US – Oligohydramnios, enlarged kidneys • IVP – Medullary streaking (Sunburst pattern) – due to dilated collecting tubules – Dense nephrogram • US – microcysts • Liver biopsy if necessary • Detailed family history • Genetic counselling
MEDULARY SPONGE KIDNEY • • • 0. 5 to 1% of all IVP Male and female affected equally Dilatation in collecting ducts In 70% bilateral renal involvement It presents in third or fourth decade with: kidney stone infection hematuria • Diagnosis with: IVP • Renal function is normally preserved
• Relatively common and benign, present at birth and not usually dx’d until age 40 -60 • Characterized by widening and cystic dilatation of distal collecting tubules • 70% bilateral, maybe affecting all papillae • Infection and calculi occasionally seen as result of urinary stasis in the tubules
• Pts present with: – Hematuria – Recurrent UTIs – Nephrolithiasis • UA may show decreased urinary concentrating ability (due to tubule damage)
• Treatment – No known therapy – Increase fluids to prevent stone formation – Treatment directed toward complications • Pyelonephritis • Renal calculi • Only small percentage of pts develop complications…overall prognosis is good
Medullary Cystic Disease • A rare, familial dz that may become symptomatic during adolescence • Manifested by many small cysts scattered through the renal medulla • Pts may present with pallor, polyuria, and lethargy • May later develop HTN
Labs • – UA shows inability to concentrate urine – CBC to confirm anemia – Chem panel to check phosphate, sodium, BUN, creatinine levels • Rad – US and CT scan to dx
• Tx – No current medical therapy to prevent progression to ESRD – Adequate water and salt replacement essential to replenish renal losses – Renal transplantation
Liddle syndrome • Rare autosomal dominant disorder • Presented with: hypertension hypokalemia metabolic alkalosis • Renin and aldostrone are suppressed • Caused by activating mutations in amiloride-sensetive sodium channel
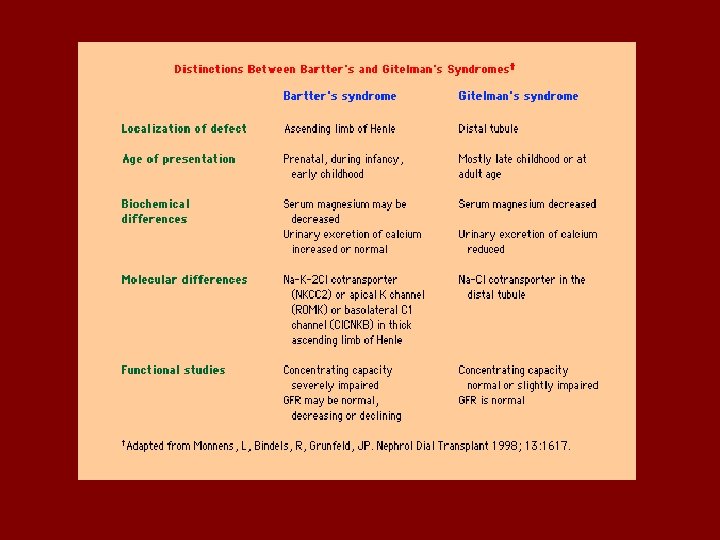
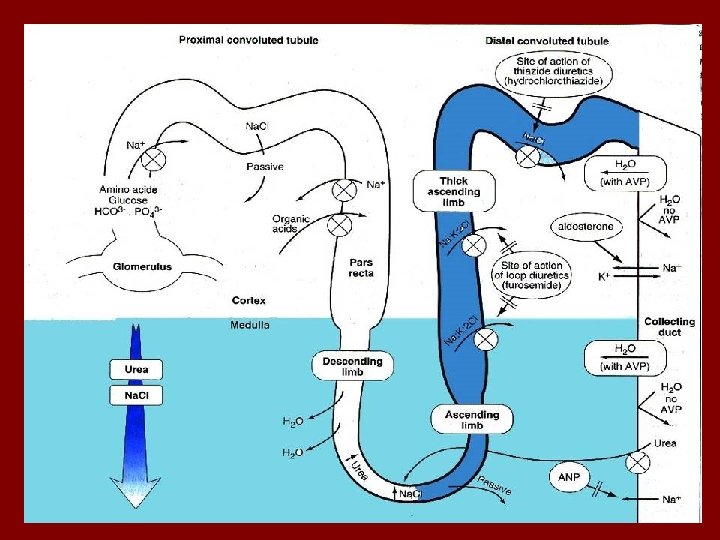
Bartter’s syndrome • Bartter’s syndrome is usually diagnosed in childhood, sometimes associated with growth and mental retardation. The defect is impaired Na. Cl reabsorption in the loop of Henle. Findings are similar to administration of a loop acting diuretic: – Salt loss leading to volume depletion and activation of the renin-angiotensin system – Increased urinary calcium
Bartter’s syndrome • Presented with: normal blood pressure hypokalemia metabolic alkalosis • Renin and aldostrone are activated • Caused by mutation in frusomide sensitive channel • Weakness, muscle cramp, polyuria
• 3 or 4 types of Bartter’s have been identified: • Defects in the luminal Na-K-Cl transporter • Defects in the luminal potassium channel • Defects in the basolateral chloride channel
Gitelman’s syndrome • Like Bartter’s an autosomal recessive disorder, but not usually diagnosed early in life. • Findings mimic administration of a thiazide diuretic: the defect is in the Na-Cl transporter. • Patients may complain of polyuria, cramps. • They do not have hypercalciuria, but typically have low serum magnesium levels.
• Diagnosis is made by history as well as lab findings. Lab findings are indistinguishable from thiazide use: – Hypokalemia, hypomagnesemia, increased renin and aldosterone levels, decreased urinary calcium. – Genetic screening?
Gitelman’s syndrome: treatment • • • Potassium Magnesium Aldactone or amiloride ACEI’s NSAIDS of no benefit Continues to require very large doses of KCl, and is on amiloride. • Magnesium levels have consistently been low or low normal.
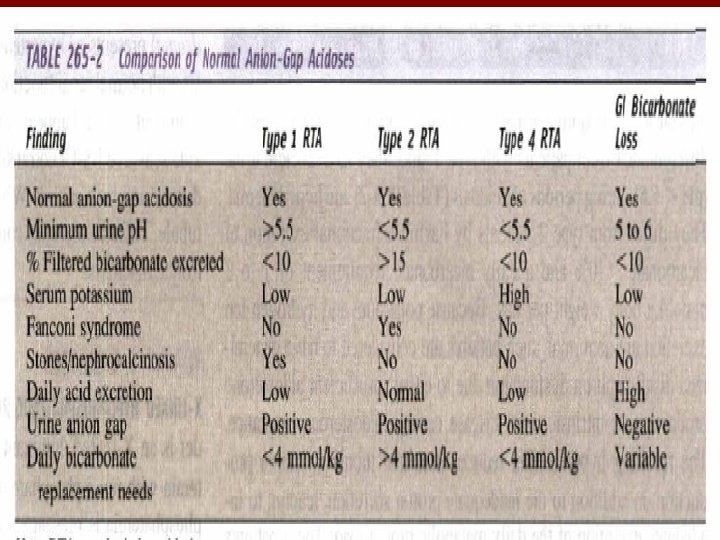
RENAL TUBULAR ACIDOSIS • Disorder of renal acidification out of proportion to the reduction in GFR • Hyperchloremic metabolic acidosis with normal serum anion gap • There are multiple forms of RTA
ALPORT SYNDROME • The most common inherited nephritis • X-linked • Defect in the gene for the a 5 chain of type IV collagen (major component of GBM) • EM: fragmentation and lamellation in GBM • Deafness in 30 to 50% • Ocular lesion in 15 to 30%
Thin segment Thick segment Note the abnormal appearance of the glomerular basement membrane (GBM) typical of Alport's syndrome, including irregular contours, areas of thinning, and marked thickening and splitting with inclusion of electron-dense granules in electron-lucent areas
BLADDER CANCER • Cancers occurred 90% in bladder, 8% in pelvic, 2% in ureter & urethra • Male/female=4/1 • Withes/ blacks=2/1 • Risk factors are: smoking aniline dyes drugs radiation infection • Symptoms: gross hematuria, irritative symptoms • Diagnosis: urine cytology, CT scan, IVP
RENAL CELL CARCINOMA • • 90% Kidney cancer Male/Female=2/1 Incidence peaks between the ages of 50 to 70 Risk factors: smoking obesity acquired cyst in ESRD tuberous sclerosis VHL
• SYMPTOMS: Hematuria abdominal pain abdominal mass fever Weight loss anemia varicocele