Imprinting Genomico Metodologie di Genetica Umana e Citogenetica











 1: 14. 500 nati vivi La sindrome di")
 Caratteristiche Cliniche: Macroglossia Difetti parete addominale Visceromegalia Gigantismo natale e")

 IGF 2, an important growth factors during embryogenesis H 19")
 IGF 2, an important growth factors during embryogenesis H 19")
 Diletta Dolfini")
")
 KCNQ 1 OT Over-espressione geni paterni Diletta Dolfini")
 Incidenza della patologia è tuttora sconosciuta, circa 1 -30/100. 000.")
 Over espressione gene H 19 e Perdita espressione IGF 2")
 Over espressione gene H 19 e Perdita espressione IGF 2")
 Over espressione gene H 19 e Perdita espressione IGF 2")
 e di Angelman (AS) Sindrome di Prader-Willi (PWS)")
 e di Angelman (AS) Sindrome di Prader-Willi (PWS)")

 Geni espressi locus materno (box neri) Geni repressi")








 SINTOMATOLOGIA: • Obesita • Abitudini alimentari eccessive • Mani e")
 SINTOMATOLOGIA: • Facies inusuale • Bassa statura • Ritardo mentale")



- amplificati peculiari per ogni")












 La riparazione delle basi male appiate")



 1. Il danno viene riconosciuto da XPC che")

 Diletta Dolfini")
 - 1/40. 000 (Giappone) •")






 ✔ Cerebellar ataxia: progressive neuronal degeneration ✔")
 Rara malattia autosomica recessiva descritta inizialmente in pazienti olandesi di")


- Slides: 72
Imprinting Genomico Metodologie di Genetica Umana e Citogenetica- Diletta Dolfini – diletta. dolfini@unimi. it
Imprinting Genomico Modello mendeliano come regola generale Trasmissione non dipendente dal genitore UGUALI PROBABILITA’DI TRASMISSIONE - Malattie a singolo gene Diletta Dolfini
Imprinting Genomico Poca attenzione verso la relazione SESSO GENITORE ESPRESSIONE O INESPRESSIONE DEL GENE NEL FIGLIO Espressione del fenotipo «condizionata» Eccezione del modello Mendeliano: Imprinting Genomico Spiegazione a tante osservazioni che contraddicono il modello mendeliano Diletta Dolfini
Imprinting impronta nel genoma Esclusione allelica: in alcuni tipi cellulari viene espresso uno solo dei due alleli, anche se entrambi sono in grado di esprimersi. Quando l’esclusione allelica è applicata selettivamente in base all’origine parentale ad un allele potenzialmente funzionale di un genitore, si parla di imprinting genomico Accade durante il periodo della sviluppo Formazione delle cellule germinali Etichette sulle informazioni genetiche Geni imprinted sono di solito coinvolti nello sviluppo Diletta Dolfini
Imprinting è un cambiamento EPIGENETICO Inattivazione del cromosoma X Differenziamento cellulare Silenziamento trasposoni Causa: alterazione della Cromatina Metilazione del DNA Modificazione covalente delle citosine, che porta all’alterazione dell’espressione del gene stesso e non alla modifica della sua sequenza nucleotidica Modificazioni post traduzionali degli Istoni Da citosina a 5 -metilcitosina Diletta Dolfini
Imprinting Cellule somatiche 2 ALLELI: NO IMPRINTING MATERNO IMPRINTING PATERNO Molti dei geni umani soggetti a imprinting si trovano raggruppati in due localizzazioni nel genoma: • una zona di circa 1 Mbp si trova sul cromosoma 11 • una zona di circa 2, 3 Mbp si trova sul cromosoma 15 Diletta Dolfini
Schema di metilazione ereditato dopo replicazione Grazie dell’esistenza di un enzima metilante, la metiltrasferasi di mantenimento (DNMT 1), una volta che uno schema di metilazione del DNA e stato stabilito, ciascun sito di metilazione viene ereditato dal DNA della progenie Diletta Dolfini
Lo schema di metilazione del filamento parentale viene ereditato immediatamente dopo la replicazione del DNA Gametogenesi Cellule Germinali Imprinting cancellato e ristabilito in base al sesso Metilazione denovo Spermatogenesi imprinting paterno Ovogenesi imprinting materno Diletta Dolfini
Imprinting Genomico Dopo la fecondazione si ha un’ondata di demetilazione del genoma, ad eccezione delle DMR (regioni con metilazione differenziale fra gli alleli parentali) In concomitanza dell’impianto il genoma dell’embrione viene rimetilato e propagato alla varie linee cellulari. Nelle cellule somatiche si assiste al mantenimento dello stato imprintato (DNMT 1) e alla lettura dell’imprinting (espressione differenziale nelle cellule somatiche). La Dnmt 1 usa come stampo il filamento parentale per la metilazione del nuovo filamento L’imprinting paterno e quello materno vengono, invece, cancellati nelle cellule primordiali germinali. Instaurazione del nuovo imprinting nelle cellule germinali in relazione al sesso dell’embrione metilazione de novo (DNMT 3 A e DNMT 3 B) Diletta Dolfini
Meccanismi di espressione differenziale metilazione diretta del promotore CH 3 competizione per l’enhancer CH 3 RNA antisenso metilazione del gene che produce un RNA antisenso CH 3
Diletta Dolfini
Cromosoma 11 Sindrome di Beckwith-Weidemann (BWS) 1: 14. 500 nati vivi La sindrome di Beckwith-Wiedemann è una sindrome da iperaccrescimento E’ correlata all’alterazione dell’imprinting genomico dei geni presenti nella regione 11 p 15 Diletta Dolfini
Sindrome di Beckwith-Weidemann (BWS) Caratteristiche Cliniche: Macroglossia Difetti parete addominale Visceromegalia Gigantismo natale e postnatale Anomalie alle orecchie Anomalie strutturali a reni e surreni Dismorfismi faciali Palatoschisi Emiperplasia (crescita asimmetrica di una o piu regioni del corpo) Occipite prominente Diletta Dolfini
GENI IMPRINTED – BWS Il cluster di geni imprinted nella regione 11 p 15 contiene almeno 12 geni in due distinti domini regolati da due imprinting centers (DMRs) Diletta Dolfini
Sindrome di Beckwith-Weidemann (BWS) IGF 2, an important growth factors during embryogenesis H 19 gene, which codes for an untranslated RNA putative tumor soppressor Diletta Dolfini
Sindrome di Beckwith-Weidemann (BWS) IGF 2, an important growth factors during embryogenesis H 19 gene, which codes for an untranslated RNA putative tumor soppressor Over-espressione geni paterni Diletta Dolfini
Sindrome di Beckwith-Weidemann (BWS) Diletta Dolfini
Sindrome di Beckwith-Weidemann (BWS)
Sindrome di Beckwith-Weidemann (BWS) KCNQ 1 OT Over-espressione geni paterni Diletta Dolfini
Sindrome di Silver-Russel (SRS) Incidenza della patologia è tuttora sconosciuta, circa 1 -30/100. 000. Maschi e femmine sono affetti in egual misura. Bambini affetti da sindrome di Silver-Russell nascono a termine con basso peso gestazionale Le principali caratteristiche craniofacciali sono rappresentate da: macrocefalia relativa con sproporzione tra neurocranio e splancnocranio, volto piccolo e triangolare fronte prominente frequente il riscontro di asimmetria a carico di un emisoma o di un arto, clinodattilia del 5º dito sindattilia delle dita dei piedi Possibile la presenza di difetti cardiaci, renali, palatoschisi, criptorchidismo, pubertà precoce. Lo sviluppo psico-motorio è nella norma nella maggioranza dei casi. Diletta Dolfini
Sindrome di Silver-Russel (SRS) Over espressione gene H 19 e Perdita espressione IGF 2
Sindrome di Silver-Russel (SRS) Over espressione gene H 19 e Perdita espressione IGF 2 Over espressione geni KCNQ 1 CDKN 1 C (geni materni)
Sindrome di Silver-Russel (SRS) Over espressione gene H 19 e Perdita espressione IGF 2 Over espressione geni KCNQ 1 CDKN 1 C (geni materni)
Cromosoma 15 Sindrome di Prader-Willi (PWS) e di Angelman (AS) Sindrome di Prader-Willi (PWS) - malattia dovuta ad assenza della funzione del ‘gene’ PWS soggetto ad imprinting materno (è espressa solo la copia fornita dal padre) che mappa in 15 q 11 -13 Sindrome di Angelman (AS) - malattia dovuta ad assenza della funzione del gene UBE 3 A, gene soggetto ad imprinting paterno (è espressa solo la copia fornita dalla madre) che mappa in 15 q 11 -13 I primi la individuarono nel 1956: Andrea Prader, Heinrich Willi e Guido Fanconi in Svizzera Diletta Dolfini
Cromosoma 15 Sindrome di Prader-Willi (PWS) e di Angelman (AS) Sindrome di Prader-Willi (PWS) - malattia dovuta ad assenza della funzione del ‘gene’ PWS soggetto ad imprinting materno (è espressa solo la copia fornita dal padre) che mappa in 15 q 11 -13 Sindrome di Angelman (AS) - malattia dovuta ad assenza della funzione del gene UBE 3 A, gene soggetto ad imprinting paterno (è espressa solo la copia fornita dalla madre) che mappa in 15 q 11 -13 I primi la individuarono nel 1956: Andrea Prader, Heinrich Willi e Guido Fanconi in Svizzera Entrambe le malattie possono essere dovute a: 1. delezione dell’intera regione cromosomica 15 q 11 -13 (75%); 2. disomia uniparentale (UPD) (materna nella PWS (25%), paterna nella AS (5%)); 3. errore di imprinting 4. solo per la sindrome di Angelman: mutazione nella copia materna del gene UBE 3 A(UBE 3 A espresso nel cervello)(15%)) Diletta Dolfini
Imprinting Genomico Geni che codificano per proteine importanti nel tessuto del cervello UBE 3 A (E 3 ubiquitin ligasi) importante durante lo sviluppo del sistema nervoso "PWS paternal-only expressed region" contiene 5 polypeptide-coding genes (MKRN 3, MAGEL 2, NECDIN, SNURF-SNRPN); NPAP 1 (gene intronless espresso in modo biallelico nei testicoli, espresso solo a livello paterno nel cervello C 15 ORF 2); un cluster di small nucleolar RNA genes (sno. RNAs) e diversi RNA antisenso (incluso quello per UBE 3 A). Diletta Dolfini
Geni espressi locus paterno (box grigi) Geni espressi locus materno (box neri) Geni repressi locus materno (box bianchi) Geni espressi in modo biallelico (box grigi scuri) AS-IC (triangle) and PWS-IC (ellipse) are shaded depending on histone modification in the area. AS-IC is dormant (gray triangle) on the paternal chromosome, whereas, on the maternal chromosome, it is acetylated and methylated at H 3 -lys 4 (green triangle), thus active. PWS-IC is active on the paternal chromosome (green ellipse), since it is also acetylated and methylated at H 3 -lys 4. However, PWS-IC at the maternal chromosome is methylated at H 3 -lys 9 and repressed (red ellipse). Differentially, the Cp. G methylated region (differentially methylated region 1 [DMR 1]) in small nuclear ribonucleoprotein polypeptide N (SNRPN) exon 1 partially overlaps with PWS-IC. Note that DMR 1 on the maternal but not paternal chromosome is methylated (black pin). Ubiquitin protein ligase E 3 A antisense transcript (UBE 3 A-ATS) originating upstream of SNRPN can either be a degradable complex with UBE 3 A transcript or prevent the extension of the ubiquitin protein ligase E 3 A (UBE 3 A) transcript (collision or upstream histone modifications represented by “X”). Neural Plast. 2012; 2012: 710943. 137 Diletta Dolfini
Perdita espressione geni materni Perdita espressione geni paterni
Pattern di espressione nel soggetto normale: sono espressi il ‘gene’ PWS del cromosoma paterno ed il gene UBE 3 A del cromosoma materno Diletta Dolfini
Pattern di espressione nel soggetto normale: sono espressi il ‘gene’ PWS del cromosoma paterno ed il gene UBE 3 A del cromosoma materno Delezioni La delezione è sul cromosoma Paterno assenza della funzione del ‘gene’ PWS, si ha Sindrome di Prader-Willi Diletta Dolfini
Pattern di espressione nel soggetto normale: sono espressi il ‘gene’ PWS del cromosoma paterno ed il gene UBE 3 A del cromosoma materno Delezioni La delezione è sul cromosoma Paterno assenza della funzione del ‘gene’ PWS, si ha Sindrome di Prader-Willi La delezione è sul cromosoma Materno assenza della funzione del gene UBE 3 A, si ha Sindrome di Angelman Diletta Dolfini
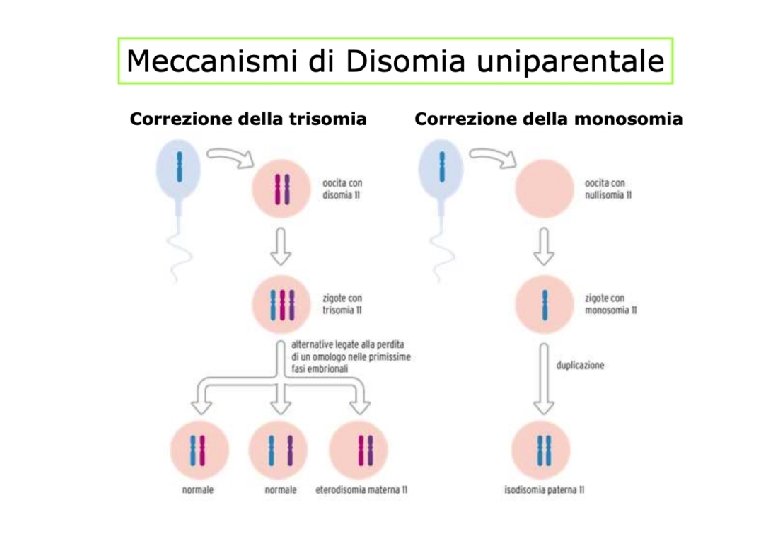
Meccanismo di disomia uniparentale Pattern di espressione nel soggetto normale: sono espressi il ‘gene’ PWS del cromosoma paterno ed il gene UBE 3 A del cromosoma materno Disomia Uniparentale Paterna (UPD) assenza funzionale del gene UBE 3 A Sindrome di Angelman UPD Materna assenza funzionale del ‘gene’ PWS Sindrome di Prader-Willi Diletta Dolfini
Pattern di espressione nel soggetto normale: sono espressi il ‘gene’ PWS del cromosoma paterno ed il gene AS del cromosoma materno mutazione nel centro di imprinting sul cromosoma P viene trasmesso con un’impronta di tipo Materno assenza funzionale del gene PWS Sindrome di Prader. Willi Diletta Dolfini
Pattern di espressione nel soggetto normale: sono espressi il ‘gene’ PWS del cromosoma paterno ed il gene AS del cromosoma materno mutazione nel centro di imprinting sul cromosoma M viene trasmesso con un’impronta di tipo Paterno assenza funzionale del gene UBE 3 A Sindrome di Angelman Diletta Dolfini
Pattern di espressione nel soggetto normale: sono espressi il ‘gene’ PWS del cromosoma paterno ed il gene AS del cromosoma materno mutazione nel gene UBE 3 A assenza funzionale del gene UBE 3 A Sindrome di Angelman Diletta Dolfini
SINDROME DI PRADER-WILLI (PWS) SINTOMATOLOGIA: • Obesita • Abitudini alimentari eccessive • Mani e piedi piccoli • Bassa statura • Ipogonadismo • Ritardo mentale Manca il contributo genetico paterno Diletta Dolfini
SINDROME DI ANGELMAN (AS) SINTOMATOLOGIA: • Facies inusuale • Bassa statura • Ritardo mentale severo • Spasticita • Convulsioni Delezione della stessa regione del cromosoma 15 ereditato dalla madre Disomia uniparentale del cromosoma 15 paterno Manca il contributo genetico materno Diletta Dolfini
Imprinting Genomico Diletta Dolfini
PSEUDOIPOPARATIROIDISMO di TIPO 1 corpo non risponde all’ormone paratiroideo chr 20 Metilazione IC (NESPAS e EXON 1 A) sul locus materno Metilazione IC (Nesp) sul locus paterno lnc. RNA(NESPAS and EXON 1) protein (GNAS e GNASxl) GNAS gene importante nella cascata di traduzione del segnale dei recettori transmembrana espresso da entrambi ma sull’allele paterno interferisce la trascrizione del lnc. RNA For the Gnas locus the alternatively spliced coding transcripts and proteins are named above and below the alleles. DMRs at Nespas and Exon 1 A (EXON A/B in human) are established in the maternal germline, while methylation at Nesp occurs during early embryonic development. Gnas is expressed biallelically, but is silenced on the paternal allele in some tissues (hatched box), in which Exon 1 A shows comparatively high expression levels (interrupted undulating line). Nesp represents a coding transcript (ORF limited to Nesp exon 2), but a regulatory RNA is initiated from a separate promoter in oocytes (red undulating line). Nespas is expressed from the unmethylated Diletta Dolfini imprinting control region (ICR) of
Diletta Dolfini
UPD- PCR FLUORESCENTI che amplificano regioni STR (Single tandem repeat)- amplificati peculiari per ogni individuo Diletta Dolfini
IL DANNO AL DNA Le mutazioni al DNA possono essere il risultato di danno al DNA non riparato o mal riparato Riparazione del danno protegge il genoma Originano la diversità genica Cause di mutazioni Errori naturali endogeni al DNA Danni esogeni al DNA (agenti fisici o chimici) Riparazione del DNA errata o inefficiente Diletta Dolfini
Meccanismi di riparazione del DNA I danni al DNA sono una continua minaccia alla stabilita genomica di una cellula. Gli organismi viventi hanno evoluto vari meccanismi per proteggere l’integrita del DNA Questi meccanismi si basano sulla attivita degli enzimi della riparazione del DNA. I molteplici sistemi di riparazione del DNA si sono evoluti per salvaguardare l’integrita dell’informazione genetica negli organismi viventi. Ogni via di riparazione e specializzata nel correggere specifici tipi di danno al DNA Esistono numerosi tipi di lesioni, un singolo processo di riparazione non potrebbe fare fronte a tutti i tipi di danno. L’evoluzione ha modulato una grande varieta di sofisticati sistemi di riparazione che riescono a recuperare la maggioranza degli insulti inflitti all’informazione genetica cellulare I sistemi di riparazione del DNA devono essersi sviluppati molto presto nell’evoluzione, e per questo sono sistemi altamente conservati Diletta Dolfini
Diletta Dolfini
Alchilazione per effetto di agenti alchilanti come le nitrosammine metilguanina si accoppia in modo sbagliato con T Analoghi delle basi azotate, come il 5 bromouracile, un analogo della timina, che però si può accoppiare a G Le distorsioni strutturali (UV) possono impedire la trascrizione e la replicazione bloccando il movimento delle polimerasi Ossidazione: agenti ossidanti (radiazionizzanti od agenti chimici che generano radicali liberi). G diventa Oxo. G che si puo accoppiare con A Diletta Dolfini
DNA damage can be caused by various genotoxic agents, such as Reactive Oxygen Species (ROS) produced during cellular metabolism, Alkylating Agents that find application in cancer therapy, Ionizing radiation (IR), which is used for radio therapy, or Ultra. Violet (UV) irradiation presenting a daily threat as it is contained in sunlight. The inflicted lesions are just as diverse, since - ROS usually lead to base modifications; - alkylating agents form adducts, while - bifunctional alkylating agents crosslink DNA to form Interstrand Cross. Links (ICLs). - IR typically induces Double-Strand Breaks (DSBs), and - UV light triggers the formation of Cyclobutane Pyrimidine Dimers (CPDs) and 6, 4 -Photo. Products (6, 4 -PPs). Cells have a repertoire to sense the different lesions and subsequently activate DNA damage checkpoint proteins. Ultimately, cells respond to the DNA damage by chromatin remodeling, modified transcription, fine-tuning of energy metabolism, cell cycle arrest, activation of DNA repair pathways and in case of irreparable damage load, induction of senescence or apoptosis. Diletta Dolfini
Scopo principale della risposta al danno al DNA: Prevenire la replicazione del DNA in presenza di un danno al DNA e la conseguente instabilità genomica 1_Attivazione dei checkpoint cellulari per arrestare il ciclo cellulare 2_Attivazione dei meccanismi di riparo del DNA 3_Riparo del danno o risposta apoptotica Diletta Dolfini
RISPOSTE CELLULARI AL DANNO AL DNA: I MECCANISMI DI RIPARAZIONE • RIPARAZIONE DIRETTA • ESCISSIONE DELLE LESIONI • excisione di basi (BER) • excisione di nucleotidi (NER) • RIPARAZIONE DEGLI ERRORI DI APPAIAMENTO TRA LE BASI (MMR) • RIPARAZIONE RICOMBINATIVA • Ricombinazione omologa (HR) • Ricombinazione non omologa (NHEJ) Diletta Dolfini
Direct reversal: a differenza delle altre risposte non richiede l’attivazione di complessi multiproteici. Un esempio: O 6 -alkylguanina. L’alchilazione impedisce l’appaiamento con la citosina. Viene rimossa grazie all’azione di un singolo enzima, Ada in E. coli corrispondente a O 6 -methyltransferase, MGMT in cellule di mammifero. MMR pathway: Mismatch Rapair, presente in eucarioti e procarioti viene utilizzata per il riparo di mismatches, piccole inserzioni o delezioni. NER pathway: Nucleotide Excision Repair , presente in eucarioti e procarioti viene utilizzata per il riparo di diverse lesioni tra cui lesioni causate da UV, mutageni chimici e chemioterapici che inducanola formazione di dimeri di pirimidina. BER pathway: Base Excision Repair, viene utilizzata in procariotied eucarioti per rimuovere basi danneggiate. Diletta Dolfini
HR pathway: Homologous Recombination, viene utilizzata in procarioti ed eucarioti per risolvere rotture a doppio filamento (DSB). Viene utilizzata in circa il 90% dei casi di DSBs nei batteri ed in lievito, e solo nel 10% dei casi di DSBs in cellule di mammifero, in cui si preferisce l’attivazione della NHEJ. Richiede l’attivazione di una serie di complessi proteici e di una cascata di eventi di trasduzione del segnale. E’ molto accurata NHEJ: Non Homologous End Joining, viene utilizzata in procarioti ed eucarioti per risolvere rotture a doppio filamento (DSB). Viene utilizzata solo in circa il 10% dei casi di DSBs nei batteri ed in lievito, e nel 90% dei casi di DSBs in cellule di mammifero. Richiede l’attivazione di una serie di complessi proteici e di una cascata di eventi di trasduzione del segnale. Non e’ molto accurata Diletta Dolfini
Diletta Dolfini
RIPARAZIONE DEGLI ERRORI DI APPAIAMENTO TRA LE BASI In un primo ciclo di replicazione l’erronea incorporazione di una base puo introdurre una mutazione. Se l’appaiamento errato sfugge al sistema di correzione di bozze da parte dell’attivita esonucleasica 3’-5’ delle DNA polimerasi replicative (aumenta la fedelta della replicazione di un fattore 100), in un secondo ciclo replicativo, la mutazione viene definitivamente fissata nella sequenza di DNA. Diletta Dolfini
BER Base Excision Repair Il meccanismo enzimatico parte dall'attivazione di una DNA-glicosilasi che riconosce la base alterata e rompe il legame N-glicosidico, poi una AP-endonucleasi 1 (AP: sito a-purinico o apirimidinico, più in generale noto come sito abasico) elimina la base azotata, lasciando il fosfato e il deossiribosio; ancora, una liasi toglie fosfato e zucchero così che una DNA-polimerasi leghi il nuovo nucleotide e la ligasi lo incorpori nel filamento. Il BER quindi può riparare la deaminazione della Citosina in Uracile o la trasformazione della Guanina in 8 -oxo-guanina, analogo dell'adenina. Diletta Dolfini
Mismatch Repair Riparazione delle basi male appaiate (MMR) La riparazione delle basi male appiate dipende da numerosi fattori presenti nelle cellule umane, fra cui Mut. S /MSH(eterodimero) o Mut. S, Mut. L / MLH(eterodimero), esonucleasi EXO 1, DNA pol. , PCNA, caricatore della pinza (RFC), proteina che lega il singolo filamento (RPA), e proteina cromosomica non istonica. HMGB 1. Il complesso Mut. S-Mut. L sul DNA con appaiamento sbagliato scatena l’attivazione del macchinario di riparazione. Mismatch Repair (MMR), che corregge errori di replicazione e di ricombinazione genetica che determinano la formazione di nucleotidi male appaiati in seguito alla replicazione del DNA. Diletta Dolfini
• L’eterodimero h. MSH 2/6 riconosce i mismatch e i loops a singola base • L’eterodimero h. MSH 2/3 riconosce i loops da inserzione/delezione • h. Mut. La e h. Mut. Lb interagiscono con MSH e con i fattori di replicazione • Sembra che i frammenti di Okazaki forniscano il segnale di discriminazione tra elica parentale e figlia Difetti nel MMR nei mammiferi predispongono al cancro, principalmente al cancro del colon, ma anche a cancro uterino, ovarico e gastrico Diletta Dolfini
CANCRO COLON-RETTALE EREDITARIO L’ 80% dei cancri del colon è sporadico, mentre un 20% ha una suscettibilità ereditaria alla malattia. Il cancro colon-rettale non associato a poliposi (HPNCC) è la forma ereditaria più comune di cancro colonrettale E’ una condizione autosomica dominante dovuta a mutazioni in uno dei diversi geni della riparazione delle basi male appaiate. Le mutazioni nei geni Mut. S o Mut. L assommano a più del 90% delle mutazioni nelle famiglie con HPNCC. Diletta Dolfini
Sindrome di Lynch I: (carcinoma ereditario colon-rettale non poliposico, Hereditary Non-Polyposis Colon Cancer – HNPCC) E una forma ereditaria di tumore al colon con trasmissione dominante, il che significa che ha una probabilita del 50% di manifestarsi nei figli di chi ne e affetto. Diversamente dalla poliposi adenomatosa familiare, la predisposizione allo sviluppo della malattia non si manifesta con la comparsa di polipi, ma direttamente con lo sviluppo del cancro al colon, in genere intorno ai 45 anni di eta. Sindrome di Lynch di tipo II: oltre al tumore al colon, comprende altre possibili neoplasie a livello: dell'endometrio, dell'ovaio, dello stomaco, del tratto urinario, dei dotti biliari. Diletta Dolfini
NER nei mammiferi (nucleotide excision repair) 1. Il danno viene riconosciuto da XPC che e legato ad h. HR 23 B 2. Successivamente si legano XPA, RPA, TFIIH e XPG. Due DNA elicasi che costituiscono TFIIH formano una bolla nel DNA 3. Si lega ERCC 1 -XPF 4. L’endonucleasi XPG taglia l’elica danneggiata al 3’ del sito di danno. ERCC 1 -XPF taglia l’elica al 5’ del sito di danno (incisione bimodale) generanado un frammento di 27 -30 nt 5. Escissione del frammento, sintesi del DNA e ligazione I geni che codificano XPA, B, C, E, F e G sono mutati nello Xeroderma Pigmentoso Diletta Dolfini
Transcription Coupled Repair TCR in mammifero ⇒ Formazione di un grosso complesso proteico insieme alla RNA polimerasi in stallo (complesso TCNER). ⇒ Il complesso disloca la polimerasi favorendo l’accesso delle proteine richieste per il BER o per il NER. ⇒ Dopo la riparazione, la trascrizione riparte. I geni che codificano CSA e CSB sono mutati nella sindrome di Cockayne (CS) (fotosensibilita della pelle) Diletta Dolfini
Malattie umane causate da alterazioni nella riparazione per escissione di nucleotidi (NER) Diletta Dolfini
Xeroderma Pigmentosum Autosomica recessiva. • 1/250. 000 (Western countries) - 1/40. 000 (Giappone) • Sviluppo precoce di carcinoma cutaneo e delle cellule squamose, melanomi, angiomi e sarcomi (50% dei pazienti) • Degenerazione neuronale (20%) • Difetti principalmente nella riparazione per escissione dei nucleotidi (NER). Mutazioni nei geni: XPA, B, C, E, F e G Diletta Dolfini
Sindrome di Cockayne • Malattia autosomica recessiva • E caratterizzata da: sensibilita alla luce, nanismo, microcefalia, difetti neuronali, sordita , invecchiamento precoce. • Non presenta predisposizione al cancro • Difetti principalmente in TCR - Mutazioni nei geni CSA e CSB - Diletta Dolfini
Riparazione delle rotture DSB NHEJ: • Predominante nei mammiferi • Riparazione inaccurata • Veloce meccanismo di emergenza • Non dipende dai cromatidi fratelli • Attiva in G 1 e sia in cellule aploidi che diploidi HR: • Prevale nelle cellule in fase S e G 2 • Riparazione accurata • Veloce meccanismo di emergenza Diletta Dolfini
Una serie di step sono alla base della risposta ai DSB • Devono essere percepiti da una molecola che e in grado di riconoscere le lesioni sul DNA o le alterazioni cromatiniche derivano dalla rottura del DNA • Le estremita rotte devono essere quindi processate • Vengono attivati i fattori di trasduzione del segnale, probabilmente attraverso modificazioni post-trasduzionali • Il segnale viene trasmesso ai fattori “effettori” • Il trasduttore iniziale e principale e ATM Diletta Dolfini
ATM = ataxia-telangiectasiamutated ➣ Serina-treonina chinasi nucleare attivata da rotture a doppio filamento ➣ Fosforila e attiva numerosi substrati che si accumulano nei “foci” di riparo (quindi associati alla riparazione del danno) o bloccano il ciclo cellulare: p 53, BRCA 1. FANC D 2, NBS 1, istone H 2 AX map of ataxia-telangiectasia-mutated controlled signalling pathways induced in response to DNA double-stranded breaks Diletta Dolfini
Model of the double-stranded-break response cycle. ATM è presente nella cellula come dimero inattivo. In seguito a danno del DNA ATM subisce un cambio conformazionale che ne induce l’attività chinasica e l’autofosforilazione in Ser 1981 che determina il rilascio dal dimero di due molecole di monomero autofosforilate ed attive (A) Undamaged section of a chromosome, showing two chromatin loops and inactive ATM dimer. (B, C) Induction of a DNA double- stranded- break (DSB), modification of ATM and recruitment of both ATM and MRE 1/RAD 50/NBS 1 (MRN). The possibility that MRN binds before ATM is shown, but the exact order is unknown. The thin black line indicates modified chromatin. (D, E) A wave of H 2 AX phosphorylation is followed by recruitment of mediators (mediator of DNA damage checkpoint protein 1 (MDC 1), p 53 -binding protein 1 (53 BP 1) and breast-cancer- associated protein 1 (BRCA 1)) to the The molecular architecture of the focus is unknown. Diletta Dolfini
L’attivazione dei checkpoints e necessaria per la percezione del danno e l’amplificazione del segnale in modo da attivare un’appropriata risposta biologica Diletta Dolfini
Clinical phenotypes of ataxia-telangiectasia (ATM gene mutated) ✔ Cerebellar ataxia: progressive neuronal degeneration ✔ Immunological dysfunction: low Ig and Ig. E; normal V(D)J recombination ✔ Cancer predisposition: lymphoma and leukaemia; chromosomal translocation ✔ Hypogonadism ✔ Sensitivity to ionizing radiation ✔ Premature aging ✔ Increased alpha-fetoprotein ✔ Small stature ✔ Insulin resistance Cellular phenotype DNA damage and repair: chromosomal instability; cell-cycle checkpoint defects in G 1, S (radioresistant DNA synthesis) and G 2/M; sensitivity to ionizing radiation, increased chromosomal breakages, telomere end-to-end fusions; DNA repair; subtle, but normal kinetics Other abnormalities: cytoskeletal defects, abnormalities in the plasma membrane, an increased number of lysosomes; high levels of trophic factors needed for growth; defects in intracellular signalling. Diletta Dolfini
Nijmegen Breakage Syndrome (NBS) Rara malattia autosomica recessiva descritta inizialmente in pazienti olandesi di origine slava Mutazione fondatrice 657 delta 5 nel gene NBS 1 Frequenza 1/100 000 Frequenza eterozigosi in individui slavi 1/155 (asintomatici) Caratteristiche fenotipiche: Fronte ristretta Viso allungato Mandibola rientrata Microcefalia Orecchie pronunciate Diletta Dolfini
Cross-talk tra le sindromi della Riparazione del DNA Link funzionali tra pathway di signalling apparentemente separate che possono convergere tutte verso una unica risposta globale al danno del DNA Diletta Dolfini
Diletta Dolfini