IL SANGUE ED I SUOI COMPONENTI Dott E



, dei globuli bianchi (leucociti)")

 sono le cellule più numerose del sangue")






: G-CSF: per i granulociti. GM-CSF: principalmente per i")






: della gittata cardiaca")











, la gravidanza (0,")

, perdita")



 (rare)")


 Emolisi: riduzione del tempo di sopravvivenza degli")


: l’ipersplenismo è caratterizzato da un aumento di")
: L’anemia emolitica autoimmune (AEA) è")
 da trauma (Anemia emolitica I GR")
.")


 normale e mutante (cellule falciformi, Emoglobina S)")


, dell’Hb (>18 g/ml) e dell’ematocrito (>50%).")
: malattia mieloproliferativa cronica ad")










: cause fisiologiche: neonati, stress, sovraffaticamento, gravidanza infezioni:")
: fase di guarigione linfocitaria/eosinofila dopo infezioni batteriche malattie allergiche")

 spesso si")







: 1. reattiva: infezioni virali (ad es. mononucleosi infettiva, rosolia,")














: è sensibile a deficit di circa il")




 Anomala generazione di fibrina nel sangue circolante.")







 Malattia caratterizzata da un aumento delle piastrine, da iperplasia megacariocitica")







- Slides: 124
IL SANGUE ED I SUOI COMPONENTI Dott. E. Li Bianchi U. O. CUBE Azienda Ospedaliera San Camillo – Forlanini Roma
SANGUE : SISTEMA BIFASICO FASE LIQUIDA PLASMA SOLUZIONE SALI MINERALI E ORGANICI FASE SOLIDA CELLULE NUCLEATE (globuli bianchi) CELLULE ANUCLEATE (globuli rossi) FRAMMENTI CITOPLASMATICI (piastrine)
Il sangue è uno dei tre compartimenti liquidi del corpo: Compartimento plasmatico: 5 litri. 45% cellule. 65% plasma: 93% acqua; 7% proteine. Compartimento intracellulare: 25 litri. Compartimento interstiziale: tra i 12 e i 15 litri (è l’unico che può oscillare). Il sangue deve essere considerato un sistema dinamico in cui le variazioni dipendono dall’equilibrio fra produzione e distribuzione dei suoi componenti. La volemia normale è di circa 70 ml/kg (pari a circa 1/14 del peso corporeo).
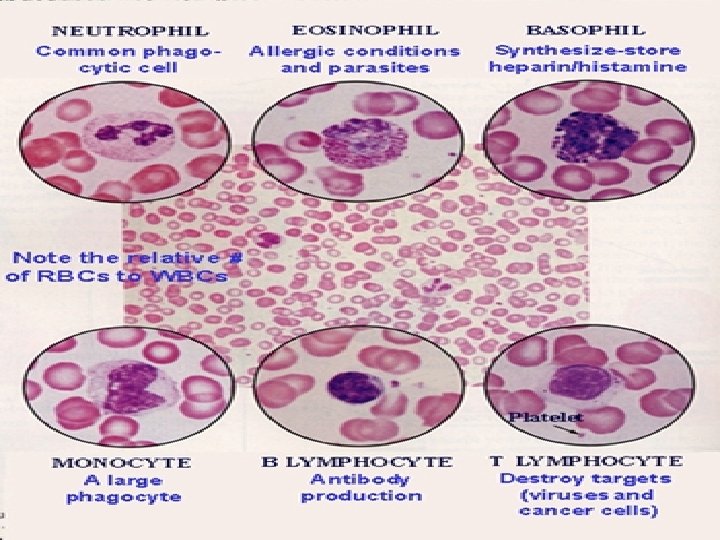
Emocromo Contempla il conteggio del numero dei globuli rossi (eritrociti), dei globuli bianchi (leucociti) e delle piastrine (trombociti), nonchè la determinazione quantitativa dell’emoglobina. Con la formula leucocitaria si intende la percentuale di ciascun tipo di globulo bianco (granulociti neutrofili, eosinofili e basofili monociti, linfociti). È detto anche esame emocromocitometrico che letteralmente significa "misurazione del colore del sangue e del numero delle sue cellule, cioè dei globuli".
Costituenti normali del sangue L’esame emocromocitometrico analizza le cellule che si trovano nel sangue, queste sono: Neutrofili: 60%; 1800 -7700/ l. Citoplasma eosinofilo, granuli fini. Nucleo polilobato, 5 -7 lobi in quelli invecchiati, a ferro di cavallo in quelli giovani. Eosinofili: 3%; 0 -450/ l. Citoplasma eosinofilo, granuli rosso scuro. Nucleo poco evidente in genere bilobato. Basofili: 1%; 0 -200/ l. Granuli azzurro scuro. Nucleo polilobato Linfociti: 30%; 1000 -4800/ l. Monociti: 4%; 0 -800/ l. Piastrine: 150000 -300000/ l. Hanno la forma di un disco di circa 23 m di diametro. Globuli rossi: 4. 500. 000 -5. 800. 000/ l nell’uomo; 4. 200. 0005. 200. 000/ l nella donna. Hanno la forma di disco biconcavo, anucleato, con un diametro di circa 7 -8 m ed uno spessore di 2. 5 m alla periferia e 0. 8 m al centro.
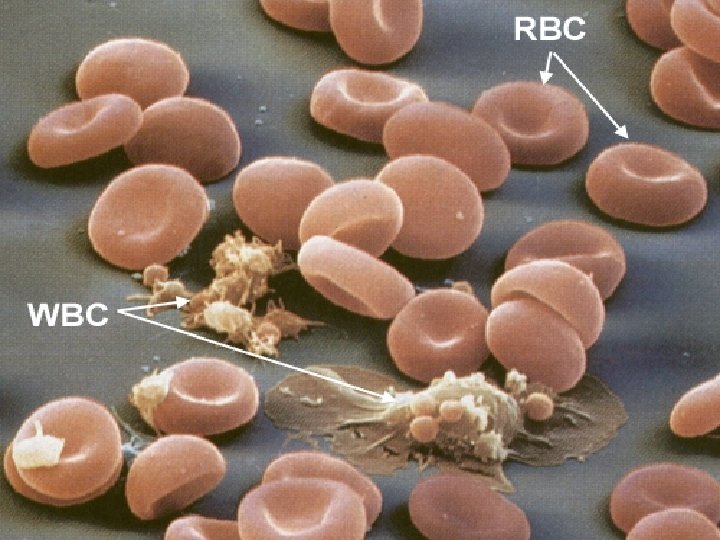
Globuli Rossi Gli eritrociti (RBC) sono le cellule più numerose del sangue
Caratteristiche dei globuli rossi: Colore: dipende dalla emoglobina. normo/ipercromico, policromico. Dimensione: normo/micro/macrocitico. Forma: normalmente è a disco biconcavo. Ogni variazione si definisce anisopoichilocitosi. La forma, l’integrità e la flessibilità della membrana sono controllate dal citoscheletro.
Misure di laboratorio: Emoglobina: e’ la proteina che trasporta l’ossigeno ed e’ presente nei globuli rossi. Il test implica la lisi degli eritrociti. Uomo: 14 -18 g/d. L; donna: 12 -16 g/d. L. Conta eritrocita ria (RBC): numero totale degli eritrociti presenti nel sangue. Ematocrito: misura del volume totale degli eritrociti in rapporto al volume totale del sangue. Uomo: 0, 42 -0, 52 L/L; donna: 0, 37 -0, 47 L/L. VES: determinazione della velocità di sedimentazione dei GR in un campione di sangue reso incoagulabile (citrato di sodio). Aumenti della VES si rilevano in caso di disprotidemia (alterazione albumina/globuline), ossia in caso di aumento, per esempio di origine infiammatoria, delle proteine di fase acuta e in caso di alterazioni qualitative e quantitative dei GR; valori più bassi si rilevano per esempio in caso di affezioni epatiche e poliglobulie.
Indici eritrocitari: MCV: volume corpuscolare medio; ematocrito/RBC. 11. 514. 5%. Indica la grandezza dei G. R. ed e’ importante per la diagnosi di anemie. MCH: emoglobina corpuscolare media (contenuto di Hb per cellula); emoglobina/RBC. 27 -31 pg. MCHC: concentrazione dell’emoglobina corpuscolare media (quantità di Hb per volume di GR); emoglobina/ematocrito. 32 -36 g/d. L. Indica se i G. R. contengono poca o molta emoglobina.
Emopoiesi L’emopoiesi è un fenomeno extravascolare che si verifica nel midollo rosso, quindi nelle ossa della cassa toracica, del cranio, del cingolo scapolare, della colonna vertebrale, del bacino e nell’epifisi delle ossa lunghe Nel midollo sono presenti due tipi di isolotti emopoietici: Un tipo è circondato dai macrofagi eritropoiesi. Un tipo è circondato da cellule reticolari mielopoiesi. Il rapporto tra i due isolotti è di 2: 1 a favore di quelli
Esiste un progenitore comune per tutte le linee cellulari: steam-cell o cellula totipotente. Da questa cellula derivano due cellule pluripotenti (committed-cell): CFU-S, cellula staminale mieloide: Eritrociti Leucociti Megacariociti CSL, cellula staminale linfoide: Linfociti T Linfociti B
Le cellule staminali possono: 1. Duplicarsi steam cell. Una stimolazione di queste cellule provoca una vera policitemia ( n cellulare). La capacità proliferativa dipende da domanda e fattori genetici. 2. Indirizzarsi commited cell (CFU). La stimolazione di una di queste cellule provoca la policitemia di una sola linea. La capacità proliferativa dipende da domanda e fattori genetici.
Fattori emopoietici (CSF: colony stimulating factors): G-CSF: per i granulociti. GM-CSF: principalmente per i granulociti ed i monociti. M-CSF: per i monociti. Eritropoietina: per gli eritrociti. Prodotta dal rene in base allo stato di ossigenazione tissutale. Essa determina: velocità di sintesi della emoglobina negli eritroblasti. sintesi proteica non emoglobinica. velocità di transito dei precursori dei GR attraverso il loro ciclo maturativo e rilasciamento in circolo.
Reticolocita rilascio della cellula nel sangue. Eritrocita maturo e mitocondri, ribosomi di perdita quindi della capacità di sintesi emoglobinica. Periodo di sviluppo normale da proeritroblasto fino al reticolocita: 5 giorni; mediante stimolazione con eritropoietina la trasformazione può avvenire in 2 giorni. Trasformazione del reticolocita in eritrocita: 1 -2 giorni. Emivita degli eritrociti: 120 giorni. Una piccola parte degli eritrociti è distrutta di solito già nel midollo osseo (= eritropoiesi inefficace).
Tra tutte le sostanze necessarie ad una normale eritropoiesi, due sono particolarmente importanti e indispensabili a tal punto che la loro carenza provoca malattia: 1. ferro, necessario per il trasporto e l’accumulo di ossigeno (la cui carenza è responsabile dell’ 80% delle anemie) 2. vitamina B 12 e acido folico, necessari per la sintesi del DNA (anemie megaloblastiche) L’eritropoiesi è regolata da un meccanismo O 2 -dipendente, tramite un fattore di crescita emopoietico, l’eritropoietina (EPO), secreta dai reni. Lo stimolo fisiologico per la formazione di eritropoietina è rappresentato dall’ipossia tissutale. L’eritropoietina, legandosi al recettore specifico presente alla superficie cellulare, promuove la differenziazione delle cellule staminali di 1° ordine ad eritroblasti nonché l’ulteriore differenziazione dell’eritropoiesi sino a reticolociti ed eritrociti.
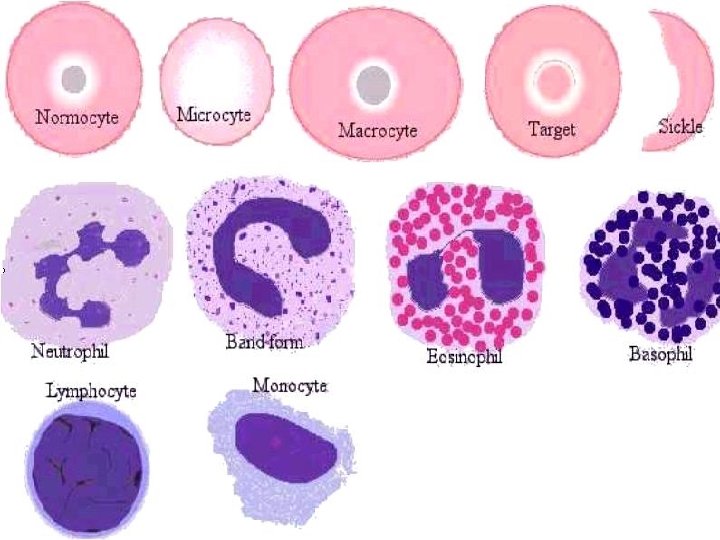
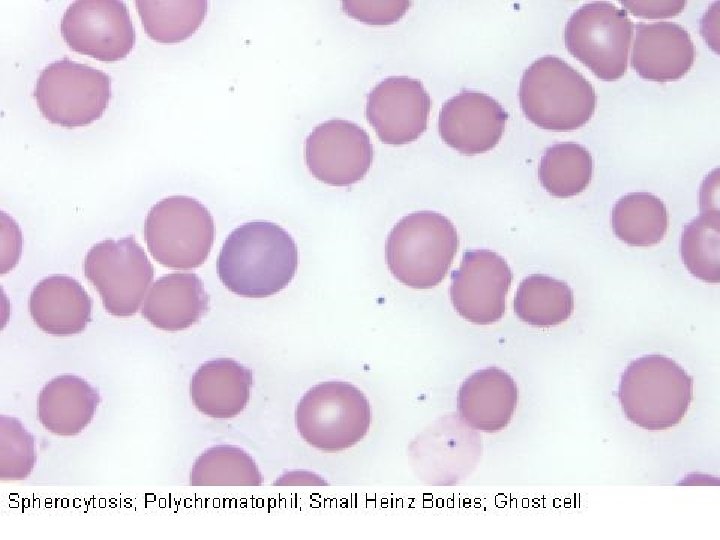
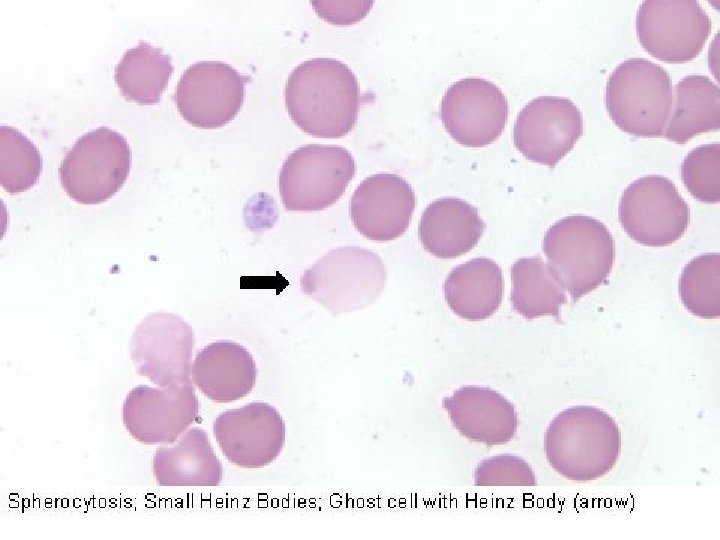
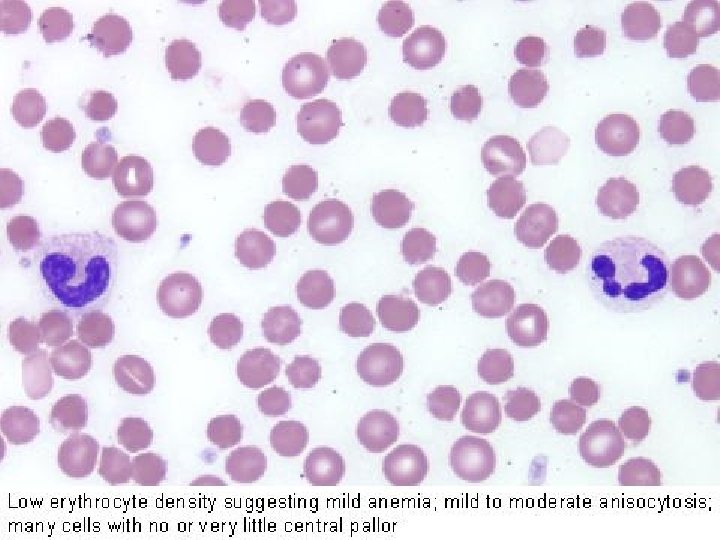
Nomenclatura morfologica degli eritrociti Anisocitosi = dimensione irregolare degli eritrociti senza modifica di forma (qualsiasi tipo di anemia) Schistociti = eritrociti rotti (ad es. Porpora trombotica trombocitopenia) Macrociti = eritrociti di forma normale ma con diametro maggiore (> 8, 5 µ) e MCV aumentato, spesso ipercromici (carenza di vitamina B 12 o acido folico) Microciti = eritrociti di forma normale ma con diametro minore (< 6, 8 µ), spesso ipocromici (anemia sideropenica) Normociti = eritrociti normali, della dimensione di 6, 8 -7, 3 µ, più chiari al centro Poichilocitosi = pronunciate modifiche di forma degli eritrociti (tutte le anemie gravi) Cellule falciformi = gli eritrociti assumono forma di falce a causa dell’emoglobina abnorme (Hb. S) (anemia falciforme) Sferociti = piccoli, discoidi, a forma rotonda, senza zona chiara centrale (sferocitosi) Cellule a bersaglio = eritrociti ipocromici con ispessimento centrale (talassemia)
Anemie Definizione Diminuita concentrazione dell’emoglobina, oppure ridotto ematocrito, oppure numero degli eritrociti al di sotto della norma: emoglobina (Hb): < 13, 5 g/dl (M) < 12, 0 g/dl (F) eritrociti: < 4, 3 milioni/µl (M) < 3, 9 milioni/µl (F) ematocrito (Ht): < 40% (M) < 37% (F) I valori di Hb e di Ht sono reciprocamente correlati e sono i parametri decisivi per la diagnosi di anemia. Il numero degli eritrociti non sempre si correla al valore di Hb e non è quindi un parametro sensibile per una definizione di anemia (ad es. anemia da carenza di ferro con Hb diminuita ma numero di eritrociti ancora normale).
Gli eritrociti più giovani contengono ancora residui di RNA sotto forma di sostanza reticolare o filiforme. Tali cellule, dette reticolociti, hanno una vita media massima di 2 giorni e rappresentano il 3 -18‰ delle emazie. La quota di reticolociti presenti nel sangue periferico e di eritroblasti nel midollo può dare indicazioni sul tipo di anemia: iperrigenerativa: reticolociti ed eritroblasti aumentati Ipo o arigenerativa: reticolociti ed eritroblasti diminuiti
L’espressione clinica dell’anemia deriva dalla ipossia tissutale ed i suoi segni peculiari rappresentano le risposte compensatorie degli apparati cardiovascolare e polmonare a tale ipossia. I sintomi clinici dipendono dalla gravità e dalla durata dell’anemia. L’anemia grave può essere accompagnata da debolezza, vertigini, cefalea, ronzii, scotomi (assenza o deficit circoscritto del campo visivo), facile stancabilità, sonnolenza, irritabilità e, nei bambini, da diminuzione delle difese immunitarie. Possono anche manifestarsi amenorrea, perdita della libido, disturbi gastrointestinali e, a volte, ittero e splenomegalia. Infine si possono avere insufficienza cardiaca o shock.
Meccanismi di adattamento (per continuare a fornire O 2 ai tessuti): della gittata cardiaca attraverso laa tachicardia. A causa dell’anemia la viscosità del sangue diminuisce ed aumenta la velocità di circolo. La pressione sistolica si mantiene su buoni valori, ma la pressione diastolica diminuisce, per cui il carico di lavoro sui ventricoli è solo leggermente aumentato. Ridistribuzione del sangue della produzione di 2 -3 DPG spostamento a dx della curva di dissociazione
Anemie da eccessiva perdita ematica Anemia postemorragica acuta: Poiché la riserva midollare è limitata, un’anemia può conseguire a una qualsiasi emorragia massiva associata alla rottura spontanea, traumatica o anche dall’incisione di un vaso sanguigno di grosso calibro, dall’erosione di un’arteria a causa di lesioni (p. es. , ulcera peptica, processo neoplastico) oppure dalla compromissione dei normali processi emostatici. Gli effetti immediati dipendono dalla durata e dall’entità dell’emorragia. La perdita improvvisa di 1/3 del volume ematico può essere fatale. Se ne può perdere fino a 2/3 senza correre tale rischio, se ciò avviene lentamente, in un periodo di 24 h. I sintomi sono causati dalla improvvisa riduzione del volume ematico e dalla successiva emodiluizione, con riduzione della capacità del sangue di trasportare l’O 2.
L’andamento dell’emorragia determina l’entità dei sintomi. Lipotimia, capogiri, sete, sudorazione, polso debole e frequente, tachipnea fanno parte del quadro. Un evento comune è l’ipotensione ortostatica. Se l’emorragia continua la PA scende a livelli critici e può sopraggiungere la morte. Durante e subito dopo l’emorragia, la conta dei GR, l’Hb e l’HCT sono falsamente alti a causa della vasocostrizione. Entro poche ore i liquidi tissutali entrano in circolo, determinando una emodiluizione e una caduta dei GR e dell’Hb proporzionali alla gravità dell’emorragia. L’anemia che ne deriva è normocitica.
Anemia postemorragica cronica: Anemia microcitica provocata da una modica ma prolungata perdita di sangue, quale si verifica a livello di una lesione emorragica cronica del tratto gastrointestinale (p. es. , ulcera peptica o emorroidi), oppure di una sede emorragica dell’apparato genitourinario. Le caratteristiche cliniche e il trattamento dell’anemia da emorragia cronica sono quelli propri dell’ anemia sideropenica.
Anemie da deficit nella produzione eritrocitaria Tra le anemie da deficit di produzione eritrocitaria tratteremo le più importanti in termini di frequenza nella popolazione generale: Anemia da carenza di ferro (sideropenica) Anemie da carenza di vitamina B 12 e acido folico (anemia megaloblastica).
Anemia sideropenica In Europa interessa circa il 10% delle donne in età fertile, più del 50% nei Paesi in via di sviluppo. È l’anemia più frequente: 80% di tutte le anemie. L’ 80% delle anemie sideropeniche si riscontra nelle donne (perché perdono Fe con la mestruazione; il fabbisogno di Fe aumenta durante la gravidanza e l’allattamento).
La carenza di ferro è la principale forma di malnutrizione nel mondo colpisce circa 1, 5 miliardi di persone. Ogni euro speso per arricchire gli alimenti di ferro ha un ritorno di 84 euro in termini di aumento della produttività e decremento degli handicap. La carenza di ferro è pericolosa soprattutto per le donne in età fertile e per i bambini, per le sue gravi conseguenze sulla salute e sulla capacità di apprendimento.
Caratteristiche di uno striscio periferico in pz con carenza di ferro
Acantociti: le forme crenate dei GR dipendono dalla osmolarità elevata del plasma che estrae acqua dalle cellule
GR ipocromici microcitici
Schematicamente, le cause di carenza di ferro sono: Carenze dietetiche (in neonati e bambini, vegetariani). Assorbimento insufficiente: acloridria, sindrome da gastroresezione, sindrome da malassorbimento. Aumento del fabbisogno (sviluppo, gravidanza, allattamento, attività sportiva). Perdita di ferro: i casi di sideropenia più frequenti (80%) sono dovuti ad emorragie croniche, tra le quali: Emorragie a livello del tratto digerente (ulcera, gastrite erosiva, emorragia da varici esofagee, ernia iatale, carcinoma, diverticolosi del colon, emorroidi, ecc. ) Metromenorragie nella donna Emorragie da interventi chirurgici o traumi
La perdita di Fe con le mestruazioni (media 0, 5 mg/die), la gravidanza (0, 5 -0, 8 mg/die), l’allattamento (0, 4 mg/die) e la perdita di sangue (da malattia, incidenti o prelievo venoso) conduce subito a una carenza di Fe, che si verifica progressivamente e culmina nelle deplezione. Stadio 1: la perdita di Fe eccede l’assorbimento, causando una progressiva deplezione dei depositi marziali (rappresentati dal contenuto del Fe midollare); Stadio 2: i depositi di Fe, ormai esauriti, non riescono più a soddisfare le richieste degli eritroblasti. La concentrazione sierica del Fe decresce, portando a una progressiva riduzione del Fe disponibile per l’eritropoiesi. Stadio 3: si instaura anemia con GR e indici in apparenza normali. Stadio 4: sono presenti microcitosi e ipocromia. Stadio 5: la carenza di Fe intacca i tessuti, determinando sintomi e segni.
In sostanza la presenza di questa patologia è indicata dai seguenti parametri di laboratorio: Hb, MCHC, MCV, quindi Microcitosi ed Ipocromia. Anisopoichilocitosi: GR a matita. Sideremia (Valori normali: 37 -147 mcg /100 ml). Ferritina: Indica la riserva organica di ferro a livello epatico. Valori inferiori alla norma indicano, con sicurezza, carenza di ferro e permettono la diagnosi differenziale tra anemia sideropenica ed anemia dovuta ad altre cause (valori normali: 20 -200 mcg/100 ml) Poiché la deficienza di Fe limita l’eritropoiesi, si manifesta reticolocitopenia
Sintomi a carico di cute e mucose: Unghie fragili, incavate e concave (coilonichia), perdita diffusa di capelli, afte orali ricorrenti, disidrosi cutanea e prurito; Sindrome di Plummer-Vinson: atrofia sideropenica della mucosa della lingua, dell’orofaringe e dell’esofago, con dolori urenti e disfagia dolorosa; Ragadi della commissura labiale. Disturbi aspecifici psichici o neurologici: Cefalea, deficit di concentrazione, irritabilità, «restless legs» , picacismo (abnorme desiderio alimentare di sostanze non commestibili). Sintomi generali dell’anemia: Pallore della cute (poco significativo) e delle mucose (più significativo); Astenia, eventuale dispnea da sforzo (diminuito apporto di O 2); Eventuale soffio sistolico (dovuto a turbolenza del flusso, da diminuita viscosità ematica).
Anemie megaloblastiche Le anemie megaloblastiche sono causate da carenza di vitamina B 12 e/o acido folico, con disturbi nella sintesi del DNA e nella maturazione del nucleo, nonché comparsa di megaloblasti. Le anemie megaloblastiche sono più frequenti per carenza di vitamina B 12. Incidenza: 9/100. 000/anno.
Anemia da carenza di vitamina B 12: macrociti spesso ovalizzati
Acido folico e vitamina B 12 rivestono particolare importanza nella sintesi dei precursori del DNA. In mancanza di B 12 non si produce DNA ma si continuare a fabbricare RNA sintesi proteica le cellule divengono sempre più grandi e si riscontrano nuclei giovani rimangono tali fino agli ultimi stadi del n. di GR 2. 000 L. Solitamente colpisce gli adulti con età compresa tra i 50 de i 60 anni. Vitamina B 12: la presenza del cosiddetto fattore intrinseco è premessa necessaria per l’assorbimento di vitamina B 12. Si tratta di un glicopeptide prodotto dalle cellule parietali della mucosa dello stomaco. La carenza di fattore intrinseco è causa di anemia megaloblastica. Acido folico: l’acido folico è contenuto negli alimenti (verdure, fegato) sotto forma di poliglutammato e viene scisso, a livello dell’intestino tenue, in monoglutammato. Tale scissione può essere disturbata dai contraccettivi orali e da alcuni farmaci.
Cause di anemia megaloblastica: Carenza di vitamina B 12 Carenze dietetiche (alimentazione vegetariana) (rare) Carenza di fattore intrinseco Esiti di gastroresezione Anemia perniciosa: caratterizzata da gastrite atrofica di tipo A con acloridria, verosimilmente dovuta alla formazione di autoanticorpi contro le cellule parietali gastriche e contro il fattore intrinseco stesso. È più frequente negli anziani, F > M. Sindrome da malassorbimento Carenza di acido folico Carenze dietetiche (alcoolismo, diete non variate in pazienti anziani) Aumentato fabbisogno (emolisi, gravidanza) Sindrome da malassorbimento Alterazioni del metabolismo dell’acido folico dovuti a particolari farmaci (difenilidantoina)
L’anemia è macrocitica, con MCV > 100 fl. È presente reticolocitopenia, a meno che il paziente non sia stato trattato. Il midollo osseo mostra iperplasia eritroide con modificazioni di tipo megaloblastico. La bilirubinemia sierica indiretta può essere elevata a causa dell’eritropoiesi inefficace e della sopravvivenza accorciata dei GR. L’analisi della vitamina B 12 sierica è il metodo più comunemente utilizzato per stabilire la carenza di B 12 come causa di megaloblastosi. Livelli < 150 pg/ml indicano con certezza una carenza di B 12.
Triade sintomatologica della carenza di vitamina B 12 Sindrome ematologica: Sintomi generali da anemia: astenia, affaticabilità, pallore. Sindrome gastrointestinale: Gastrite atrofica autoimmune di tipo A con acloridria in caso di anemia perniciosa. Alterazioni trofiche della mucosa, glossite atrofica, lingua liscia e arrossata e parestesie linguali urenti. Sindrome neurologica: Degenerazione spinale, funicolare e demielinizzazione dei cordoni posteriori (atassia spinale) e della via piramidale (paresi spastica, segni piramidali). Si manifestano sintomi e segni da polineuropatia con parestesie dolorose a carico di mani e piedi (formicolio, andatura atassica). Eventuale areflessia delle estremità inferiori, occasionalmente anche sintomi psicotici.
Anemie da eccessiva distruzione eritrocitaria (Anemie emolitiche) Emolisi: riduzione del tempo di sopravvivenza degli eritrociti dai normali 120 giorni sino a poche settimane o giorni. Si distinguono: emolisi intra- ed extravascolare. Se un concomitante incremento dell’emopoiesi mantiene costanti i livelli di emoglobina, si parla di emolisi compensata; altrimenti si parla di anemia emolitica.
Gli eritrociti più vecchi vengono rimossi dal torrente circolatorio dopo un’emivita di 120 giorni. L’ 85% di questa emolisi fisiologica avviene in sede extravascolare nel sistema reticolo-endoteliale (SRE). La sede emocateretica principale è la milza. In caso di emolisi patologica rilevante, gli eritrociti vengono distrutti anche nel fegato e nel midollo osseo. Tutte le emolisi croniche portano ad una stimolazione dell’eritropoiesi con degli aspetti tipici: nel midollo osseo: proliferazione eritroblastica con maturazione normale. Il rapporto leuco/eritroblastico si sposta a favore della serie rossa. nel sangue periferico: reticolocitosi (a condizione che il midollo sia normale).
Anemie emolitiche da difetti eritrocitari estrinseci: Nessuna anomalia dei GR può essere identificata o implicata nell’emolisi da deficit extraglobulari. La distruzione dei GR è in rapporto a circostanze estranee ai GR. Le cellule del donatore vengono distrutte a una velocità uguale a quella delle cellule autologhe.
Anemia da iperattività del SRE (ipersplenismo): l’ipersplenismo è caratterizzato da un aumento di volume della milza, accompagnato da una maggiore attività filtrante e fagocitaria a carico dei GR, leucociti e piastrine. Malattie associate a iperplasia reticoloendoteliale sono quelle maggiormente implicate nella genesi dell’ipersplenismo, che può anche derivare da qualsiasi patologia che determina splenomegalia. La splenomegalia è il segno caratteristico dell’ipersplenismo; la grandezza della milza è direttamente proporzionale al grado dell’anemia. Per ogni 2 cm di milza che supera il margine costale ci si può attendere un calo del’Hb di circa 1 g. L’anemia e le altre citopenie sono di solito modeste e asintomatiche, a meno che non coesistano altri meccanismi responsabili della loro gravità.
Anemie da alterazioni immunologiche: Anemia emolitica autoimmune (AEA): L’anemia emolitica autoimmune (AEA) è identificata dalla presenza di autoanticorpi (Ac) che reagiscono contro i GR. L’anemia emolitica da anticorpi caldi è la forma più comune di AEA. L ’anemia è di solito grave e può essere fatale. Questi anticorpi possono comparire spontaneamente, in associazione a talune malattie (LES, linfomi) o dopo stimolo da parte di un farmaco (p. es. , a-metildopa, levodopa). La malattia da anticorpo freddo (malattia da agglutinina fredda) è un’anemia emolitica causata da un autoanticorpo che reagisce a una temperature < 37°C (di solito < 30°C). La malattia è associata a infezioni (soprattutto polmoniti da micoplasma o mononucleosi) e a stati linfoproliferativi. Anemia associata a sensibilità al complemento: L’emoglobinuria parossistica notturna è un raro disordine caratterizzato da episodi di emolisi ed emoglobinuria, quest’ultima più accentuata durante il sonno. Le crisi possono essere scatenate dalle infezioni, dalla somministrazione di Fe, di vaccini o dalle mestruazioni.
Anemie da danno meccanico: Anemia emolitica microangiopatica) da trauma (Anemia emolitica I GR si rompono quando sono esposti a deformazione o turbolenza di circolo eccessive; la diagnosi è data dalla presenza di frammenti di GR di forma strana nel sangue periferico (p. es. , triangoli o forme a elmetto). Anemia emolitica causata da agenti infettivi Gli agenti infettivi possono provocare emolisi attraverso un’azione diretta delle tossine (p. es. , da Clostridium perfrigens, da streptococchi alfa o beta emolitici o da meningococchi) oppure per invasione e distruzione dei GR da parte del microrganismo (p. es. , Plasmodium e Bartonella sp).
Anemie emolitiche da difetti eritrocitari intrinseci: Anemie emolitiche da alterazioni della membrana eritrocitaria (sferocitosi). La sferocitosi ereditaria è una malattia cronica trasmessa come carattere dominante e caratterizzata da emolisi di GR sferoidali, anemia, ittero e splenomegalia. Diverse anomalie a carico delle proteine di membrana sono responsabili delle modificazioni in senso sferocitico. La riduzione dell’area di superficie della cellula si ripercuote negativamente sulla flessibilità necessaria alla cellula per passare attraverso il microcircolo splenico. Come risultato, si verifica emolisi nella milza. L’ellissocitosi ereditaria (ovalocitosi) è una rara malattia autosomica dominante nella quale i GR sono ovali o di forma ellittica o ovalare; l’emolisi è di solito assente o minima, con anemia modesta o assente. L’anomalia dei GR consegue ad alterazione delle proteine di membrana.
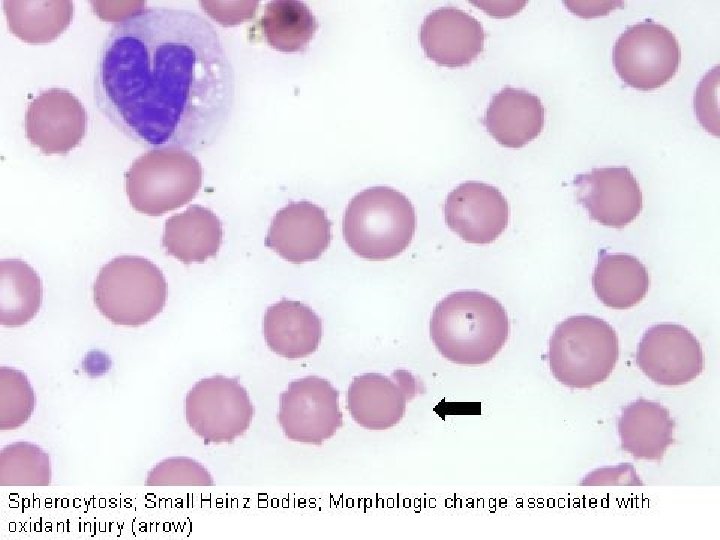
Anemie da alterazioni del metabolismo eritrocitario: Deficit dello shunt degli esosi monofosfati e del metabolismo del glutadione. L’unico deficit di rilievo in questa via metabolica è quello della glucoso-6 -fosfato deidrogenasi (G 6 PD), comunemente chiamato favismo. Deficit della glucosio 6 -fosfato deidrogenasi protezione contro gli agenti ossidanti. L’emolisi è prevalentemente intravascolare, l’Hb precipitata produce corpi di Heinz e conseguente danno eritrocitario nella microcircolazione. L’emolisi si evidenzia nella popolazione più vecchia dei GR dopo esposizione a farmaci o altre sostanze che producono perossidi e provocano l’ossidazione dell’Hb e della membrana del GR. Questi agenti comprendono i salicilati, i sulfamidici, i nitrofurani, alcuni derivati della vitamina K e, in alcuni individui di razza bianca, i semi di fava. Tuttavia gli eventi scatenanti più comuni oltre ai farmaci sono la febbre, le infezioni acute virali e batteriche e l’acidosi diabetica.
Anemie da difettosa sintesi di emoglobina. Sono alterazioni genetiche della molecola dell’Hb che si rendono evidenti attraverso modificazioni delle caratteristiche chimiche, della mobilità elettroforetica o delle proprietà fisiche della proteina. Anemia falciforme: anemia emolitica cronica ereditaria che si manifesta quasi esclusivamente nella razza nera ed è caratterizzata da GR con forma a falce (nel versante venoso), alterazione dovuta alla presenza di Hb S nell’individuo omozigote. La forma deossigenata di questa emoglobina precipita sottoforma di cristalli i quali si attorcigliano tra di loro a spirale. Cellule falciformi, elettroforesi Hb S. Talassemie o anemia Mediterranea: gruppo di anemie emolitiche croniche si manifestano in popolazioni provenienti dai paesi del Bacino Mediterraneo e dal Sud-Est Asiatico; esse sono tutte caratterizzate da una alterazione nella sintesi dell’Hb e da eritropoiesi inefficace. -talassemia, -talassemia
Cromatografia bidimensionale di (Emoglobina A) normale e mutante (cellule falciformi, Emoglobina S)
Anemia falciforme is caused by the substitution of a single nucleotide base in the gene coding for the Beta chain of hemoglobin. A single base pair substitution cause the normal aa, glutamate (hydrophilic and polar) to be replaced with valine (hydrophobic and nonpolar). Result: hemoglobin is less water -soluble, especially under acidic conditions. It crystalizes when the blood becomes acidic (as from carbonic acid, during exertion).
Emolisi croniche Emolisi compensata: la bilirubina plasmatica è ancora normale, la vita media degli eritrociti è diminuita ed è compensata dall’aumento dell’eritropoiesi (sino a 10 volte il valore normale). Anemia emolitica: l’emolisi è maggiore della capacità di compensazione del midollo: segni di emolisi con anemia. I sintomi principali di emolisi cronica conclamata sono: sintomi generali (astenia, affaticabilità, dispnea da sforzo, vertigini), eventuale ittero, splenomegalia e calcolosi biliare pigmentata. Crisi emolitica acuta febbre, brividi, eventualmente collasso ittero, iperbilirubinemia emoglobinuria con urina color birra scura: pericolo di insufficienza renale acuta tumefazione e dolore splenico, dolori lombari.
Poliglobulie Caratterizzate da dei GR (6 -10 milioni), dell’Hb (>18 g/ml) e dell’ematocrito (>50%). In condizioni normali l’eritropoiesi è regolata dall’eritropoietina (EPO), secreta a livello renale ed il cui principale stimolo alla produzione è rappresentato dalla ipossia tissutale. Un aumento della concentrazione di eritropoietina si riscontra in caso di: — carenza sistemica di O 2 (anemie, insufficienza cardiaca/polmonare) — carenza locale di O 2 (cisti renali, idronefrosi) — ipernefroma (sintesi paraneoplastica di eritropoietina). Concentrazione diminuita di eritropoietina in: — policitemia vera (proliferazione neoplastica che interessa soprattutto la linea eritroide) — insufficienza renale (anemia renale) in cui è assente l’aumento dell’eritropoietina tipico delle anemie
Policitemia assoluta Policitemia primaria (policitemia vera o morbo di Vaquez): malattia mieloproliferativa cronica ad esito infausto, caratterizzata da un’abnorme proliferazione di tutte le linee cellulari midollari, in particolare da un aumento della concentrazione dell’Hb e della massa dei GR (eritrocitosi). C’è un aumento della produzione e del turnover dei GR, dei neutrofili e delle piastrine. I disturbi (debolezza, cefalea, sensazione di testa vuota, disturbi visivi, affaticamento, dispnea) di solito possono essere attribuiti all’espansione del volume ematico e all’iperviscosità. La faccia può essere rubizza e le vene retiniche congeste. Comune è l’epatomegalia e più del 75% dei pazienti presenta splenomegalia. Infine, l’attività eritropoietica si riduce nel midollo. GB immaturi e precursori dei GR possono ritrovarsi nel sangue periferico, mentre si sviluppano marcata anisocitosi, poichilocitosi, con microciti, ellissociti e cellule a goccia. La PV deve essere considerata negli uomini con Htc > 54% e nelle donne con Htc > 49%. Senza trattamento, il 50% dei pazienti sintomatici muore entro 18 mesi dalla diagnosi.
Policitemia assoluta secondaria: è caratterizzata dall’iperproduzione di eritropoietina. Può essere la conseguenza del soggiorno ad alte quote, di malattie cardiopolmonari croniche, di shunt dx-sx, di riduzione della ventilazione alveolare, di patologie renali. Fumo: il fumo può causare un’eritrocitosi reversibile. La carbossiemoglobina è una conseguenza dell’inalazione del fumo di tabacco. L’eritrocitosi è causata dall’anossia tissutale. Ipossiemia arteriosa: i pazienti con patologia polmonare cronica o con shunt intracardiaci da destra a sinistra con ipossiemia possono sviluppare un’eritrocitosi. Anche in caso di prolungata esposizione ad alte quote o nelle sindromi da ipoventilazione centrale, può esserci un incremento della massa eritrocitaria. Emoglobinopatie con alta affinità per l’O 2 Eritrocitosi associata a tumore: i tumori renali e le cisti possono causare eritrocitosi dovuta all’aumentata secrezione di eritropoietina.
Policitemia relativa Quando l’aumento del numero di GR, dell’Hb e dell’ematocrito non si associa ad aumento della massa eritrocitaria totale, ma è dovuta ad una riduzione del volume plasmatico (poliglobulia da stress, sindrome di Gaisböck).
Poiché l’Htc è un rapporto dei GR circolanti per unità di volume di sangue intero, un Htc elevato può essere causato da una riduzione del volume plasmatico. Quindi, una diagnosi di eritrocitosi vera è basata sulla dimostrazione di una massa eritrocitaria aumentata. Nella eritrocitosi relativa (cioè, policitemia da stress, la sindrome di Gaisböck), la massa è normale e l’aumento dell’Htc è causato da una riduzione del volume plasmatico. Più comune è l’eritrocitosi secondaria a patologia polmonare, a policitemia del fumatore causata da elevati livelli di carbossiemoglobina e a tumori producenti sostanze eritropoietiche. Se la concentrazione arteriosa di O 2 è < 92%, un’ipossia tissutale può essere alla base dell’eritrocitosi. La P 50 (pressione parziale di O 2 relativa a una saturazione dell’Hb del 50%) rappresenta una valutazione dell’affinità dell’Hb per l’O 2 ed esclude un’alta affinità dell’Hb (un’anomalia familiare) quale causa di eritrocitosi. I pazienti con Policitemia Vera hanno livelli bassi o indeterminabili di eritropoietina sierica; quelli con eritrocitosi indotta da ipossia hanno livelli elevati; quelli con eritrocitosi associata a tumore hanno livelli normali o aumentati.
Malattie dei globuli bianchi I leucociti, o globuli bianchi, sono incaricati della difesa dell'organismo. La densità di leucociti nel sangue è di 4000 -10000 /mm 3. I leucociti si dividono in due categorie: granulociti e cellule linfoidi (o agranulociti). Il termine di granulociti è dovuto alla presenza di granuli nel citoplasma di queste cellule. I granulociti si distinguono in neutrofili, eosinofili (o acidofili), basofili. Le cellule linfoidi, invece, si distinguono in linfociti e monociti.
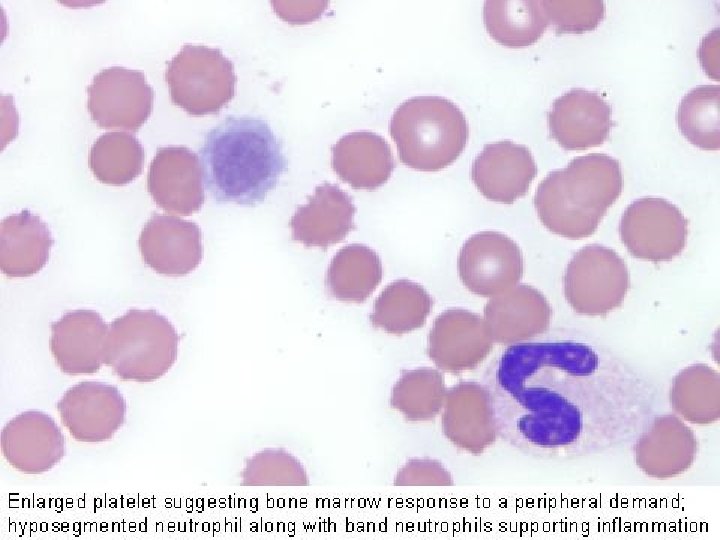
Granulociti Derivano dal midollo osseo, il loro citoplasma è ricco di granuli che assumono colorazioni caratteristiche e che ne aiutano il riconoscimento. Il nucleo è raggruppato in masserelle o lobi. Nel sangue ci sono anche cellule immature. Esse si distinguono per avere il nucleo meno segmentato.
I neutrofili sono molto attivi nel fagocitare batteri e sono presenti in grandi quantità nel pus delle ferite. I neutrofili sono i leucociti più comuni. Hanno un diametro di 12 -15 µm. Si riconoscono per il nucleo suddiviso in 2 -5 lobi. Il citoplasma è trasparente perchè ha granuli piccoli e debolmente colorati di rosa. Nel nucleo dei neutrofili delle femmine, è visibile un'appendice a forma di piccola mazza. E' il secondo cromosoma X, inattivato.
Gli eosinofili aggrediscono parassiti e fagocitano i complessi antigene-anticorpo. Gli eosinofili sono abbastanza rari nel sangue. La loro dimensione è la stessa dei neutrofili. Il nucleo è generalmente bilobato, ma sono stati osservati anche nuclei con 3 o 4 lobi. Il citoplasma è pieno di granuli che assumono un colore rosa-arancione caratteristico. Il nucleo risulta ancora ben visibile.
I basofili secernono sostanze anticoagulanti, vasodilatatrici come l'istamina e la serotonina. Anche se possiedono capacità fagocitaria, la loro funzione principale è quella di secernere sostanze che mediano la reazione di ipersensibilità. I basofili sono i leucociti più rari (meno dell'1%). Sono relativamente piccoli: 9 -10 µm di diametro. Il citoplasma è molto ricco di granuli che prendono una colorazione porpora scuro. Nei basofili, la quantità di granuli è tale da nascondere il nucleo, generalmente bi-trilobato.
I granulociti assicurano la difesa cellulare aspecifica, in particolare contro batteri e funghi. Normalmente il 90% dei granulociti si trova nel midollo osseo, solo il 2 -3% nel sangue circolante e il resto nei tessuti. Segnali umorali (ad es. interleuchina-1, fattore complementare C 3) provocano una mobilizzazione dei granulociti del midollo al sangue. Nel sangue i granulociti si dividono in un pool marginale attorno alle pareti dei vasi ed in un pool circolante nel sangue.
Modificazioni reattive Una infezione batterica acuta comporta tipiche variazioni a carico dei globuli bianchi e delle proteine sieriche.
Granulocitosi neutrofila (= neutrofili > 7. 500/µl): cause fisiologiche: neonati, stress, sovraffaticamento, gravidanza infezioni: soprattutto batteriche: rari sono i casi con quota leucocitaria normale o diminuita (tifo, brucellosi) infiammazione: febbre reumatica, connettiviti, pancreatite, ascessi neoplasie: affezioni mieloproliferative necrosi tissutali: infarto cardiaco, polmonare, ustioni ed altro disturbi metabolici: gotta, crisi tireotossica, coma diabetico e uremico farmaci: ad es. steroidi, adrenalina, litio altro: in seguito a splenectomia, coliti, emorragia acuta, emolisi acuta, traumi, shock, fumo (leucocitosi da fumo)
Eosinofilia (= eosinofili > 450/µl): fase di guarigione linfocitaria/eosinofila dopo infezioni batteriche malattie allergiche malattie parassitarie si manifestano con eosinofilia marcata (ad es. infestazioni da vermi) affezioni cutanee croniche neoplasie: ad es. malattia di Hodgkin, malattie mieloproliferative, carcinomi metastatizzati altre cause: ad es. sindrome di Churg-Strauss e vasculiti da ipersensibilità, endocardite di Löffler, malattia di Addison, ecc.
Neutropenia Si definisce leucopenia una riduzione del numero dei GB circolanti al di sotto di 4000/ml. Solitamente una leucopenia è caratterizzata da un ridotto numero di granulociti neutrofili (Granulocitopenia; agranulocitosi) La riduzione del numero dei granulociti neutrofili nel sangue comporta di solito aumentata suscettibilità alle infezioni batteriche e fungine. Una classificazione delle neutropenie può essere effettuata con la conta dei neutrofili (GB totali, percentuale dei neutrofili e dei precursori) e sul relativo rischio di infezioni: neutropenie lievi (1000 -1500/ml), moderate (500 -1000/ml) o gravi (< 500/ml). Una neutropenia acuta, grave, secondaria a ridotta produzione, spesso è pericolosa per la vita del paziente immunocompromesso.
La neutropenia acuta (che si manifesta nello spazio di pochi giorni) spesso si sviluppa quando l'utilizzo dei neutrofili è rapido e la produzione carente. La neutropenia cronica (con durata di mesi o anni) in genere deriva da una ridotta produzione o da un eccessivo sequestro splenico di neutrofili. La neutropenia può essere classificata a seconda che sia un epifenomeno di fattori estrinseci alle cellule mieloidi midollari o un difetto intrinseco presente nei progenitori mieloidi.
Neutropenia secondaria La neutropenia indotta da farmaci può essere dovuta a diversi meccanismi sottostanti (immunologici, tossici, idiosincrasici o reazioni di ipersensibilità) e deve essere differenziata dalla neutropenia grave che si sviluppa prevedibilmente dopo la somministrazione di grandi quantità di farmaci citoriduttivi antineoplastici o a seguito di radioterapia e da quella causata da infezioni virali. La carente produzione di neutrofili può avvenire quando una leucemia, un mieloma, un linfoma o tumori solidi metastatici (p. es. , della mammella, della prostata) infiltrano e sostituiscono il midollo osseo. Una neutropenia transitoria spesso accompagna le infezioni virali (p. es. , stadio iniziale della mononucleosi infettiva) e la sepsi è una causa particolarmente grave di neutropenia. Neutropenia cronica spesso accompagna l'infezione da HIV, il risultato di una carente produzione di neutrofili e di una accelerata distruzione di neutrofili da parte di anticorpi. Neutropenie autoimmuni possono associarsi alla presenza di anticorpi circolantineutrofili e possono manifestarsi isolatamente o con malattie associate.
Neutropenia causata da difetti intrinseci delle cellule mieloidi o nei loro precursori: questo tipo di neutropenia non è comune. La neutropenia ciclica è un raro disordine granulocitopoietico congenito. Può essere trasmesso in modo autosomico dominante ed è caratterizzato da irregolari, periodiche oscillazioni del numero dei neutrofili periferici. La neutropenia cronica idiopatica rappresenta un gruppo di non comuni disordini che coinvolgono cellule staminali commissionate della serie mieloide; sono presenti un normale numero di precursori dei GR e delle piastrine. Non è presente splenomegalia. Il grado di suscettibilità alle infezioni è quasi proporzionale alla conta ematica dei neutrofili nei pazienti conte assolute dei neutrofili < 500/ml.
Alcuni pazienti con neutropenia cronica conte dei neutrofili < 200/ml non hanno esperienza di molte infezioni gravi, probabilmente poiché il resto del sistema immunitario rimane intatto. Comunemente, tuttavia, i pazienti con neutropenia ciclica o neutropenia grave congenita presentano ulcere orali, stomatiti o faringite associata a ingrandimento linfonodale nel corso degli stati di grave neutropenia. Si manifestano spesso polmoniti e periodontiti croniche. I pazienti la cui neutropenia è secondaria a disordini di produzione acquisiti, derivanti da cancro o da chemioterapia, sviluppano con maggiore probabilità gravi infezioni batteriche, poiché il loro sistema immunitario è globalmente compromesso.
La diagnosi di neutropenia di solito si sospetta in un paziente con infezioni frequenti, gravi o usuali e viene confermata da basse conte ematiche. Durante l'esame obiettivo, particolare attenzione deve essere diretta ai più comuni siti primari di infezione: spesso le superfici mucose, quali il tratto alimentare (gengive, faringe), dove il danno indotto dalla chemioterapia può consentire l'invasione di organismi colonizzanti o la pelle, dove cateteri vascolari possono fungere da porta di infezione. Altri siti comuni di infezione includono i polmoni, il peritoneo, i siti di aspirazione del midollo osseo e di prelievo venoso e le unghie.
Linfociti I linfociti hanno la dimensione di 7 -15 micrometri con un nucleo rotondeggiante, un citoplasma scarso e pochi granuli. Svolgono un ruolo molto importante nel sistema immunitario: hanno il compito di reagire in modo specifico nei confronti di qualsiasi agente estraneo, o antigene. Si parla in questo caso di attivazione del linfocita e linfociti attivati si possono ritrovare in molti tessuti. I Linfociti derivano dalla cellula staminale multipotente e a seconda del luogo, all'interno dell'organismo, nel quale avviene la maturazione cellulare, si ottengono due linee linfocitarie ben distinte: i linfociti B e i linfociti T.
Linfociti T Prendono nome dal fatto che, dopo essere stati prodotti nel midollo osseo, migrano nel timo, dove avviene la loro maturazione, per cui i linfociti T imparano a riconoscere le cellule dell’organismo. I linfociti T maturi possono essere divisi in due tipi fondamentali. I linfociti T helper, che per riconoscere l’antigene devono venire in contatto con cellule macrofagiche „presentino“ loro l’antigene. Quando viene in contatto con il macrofago, il linfocita T helper può riconoscere specificamente i frammenti della struttura antigenica demolita ed esposta all’esterno: questo evento attiva il linfocita T helper, che quando reincontra la struttura antigenica secerne una inteleuchina-2. A sua volta, l’interleuchina-2 secreta dai linfociti T helper attiva quei linfociti T citotossici che abbiano anch’essi riconosciuto lo stesso antigene, inducendoli a dividersi e a dare origine a un clone di cellule citotossiche, capaci di uccidere specificamente tutte le cellule che presentano quell’antigene.
Linfociti B Nell’organismo umano, i linfociti B maturano nello stesso organo in cui nascono, il midollo osseo. In seguito alla loro maturazione passano nel circolo sanguigno, ma, essendo cellule più sedentarie dei linfociti T, tendono a localizzarsi nei linfonodi. Un linfocita B maturo viene attivato dal contatto con l’antigene che si lega specificamente alle immunoglobuline della classe Ig. M e Ig. D esposte sulla sua membrana. In seguito a questo contatto specifico, il linfocita B diviene suscettibile a una serie di molecole segnale (linfochine) rilasciate da un linfocita helper, che abbia a sua volta riconosciuto lo stesso antigene. Il legame di queste linfochine con i loro recettori specifici, espressi sulla membrana dei linfociti B, induce il linfocita B a dividersi attivamente e a generare un clone di cellule figlie. Da queste prenderanno origine due tipi di cellule: le plasmacellule: cellule a vita breve, capaci di produrre e secernere una gran quantità di anticorpi, e destinate a morire una volta cessata questa funzione; I linfociti B memoria sono cellule con una vita molto lunga, con il compito di mantenere il „ricordo“ dell’antigene che è stato riconosciuto per la prima volta dalla cellula capostipite del clone.
Linfocitosi assoluta (> 4. 000/µl): 1. reattiva: infezioni virali (ad es. mononucleosi infettiva, rosolia, ecc. ) «fase di guarigione linfocitaria» di infezioni batteriche tubercolosi, sifilide, pertosse, malattia di Bang; spesso i bambini reagiscono a varie infezioni con una linfocitosi reattiva. 2. neoplastica: leucemia linfatica cronica (LLC).
Linfocitopenia Una conta linfocitaria totale < 1000/ml negli adulti o < 3000/ml nei bambini < 2 anni. La conta linfocitaria normale negli adulti è 10004800/ml e nei bambini con meno di due anni, 3000 -9500/ml.
La linfocitopenia congenita può essere associata a malattie da immunodeficienza congenita, che possono anche avere un'anomalia quantitativa o qualitativa della cellula staminale che determina una linfocitopoiesi insufficiente. Limfocitopenia associata ad altre cause, quali la sindrome di Wiskott-Aldrich, può derivare da una accelerata distruzione di cellule T. Un meccanismo simile è presente in pazienti con deficit di adenosina- deaminasi e deficit di purinnucleoside-fosforilasi.
La linfocitopenia acquisita si riferisce alle sindromi associate a deplezione di linfociti ematici non secondaria a malattie congenite. L'AIDS è la più comune malattia infettiva associata a linfocitopenia, che deriva da distruzione dei linfociti T CD 4+ infettati dall'HIV. La linfocitopenia può anche riflettere un'insufficiente produzione e proliferazione linfocitaria derivante dalla distruzione della normale architettura timica o linfoide. Altre affezioni virali e batteriche possono associarsi a linfocitopenia. La linfocitopenia jatrogena è causata dalla chemioterapia citotossica, dalla radioterapia e dalla somministrazione di globulina antilinfocitaria. I glucocorticoidi possono causare linfocitopenia attraverso distruzione cellulare. Malattie sistemiche associate ad autoimmunità (p. es. , LES, AR, miastenia gravis) possono indurre linfocitopenia.
La linfocitopenia, in genere, non causa sintomi ed è generalmente messa in evidenza durante la diagnosi di altre affezioni, particolarmente infezioni virali, fungine o parassitarie. I pazienti linfocitopenici sperimentano infezioni ricorrenti, spesso presentano risposte inusuali ad agenti infettivi benigni o sviluppano infezioni da organismi inusuali. Una polmonite da Pneumocystis carinii, da citomegalovirus, da rosolia o da varicella suggerisce una potenziale immunodeficienza; una polmonite determinata da una di queste infezioni è spesso fatale. Questi pazienti presentano anche una più alta incidenza di tumori maligni e di disordini autoimmuni. Essi possono avere tonsille o linfonodi assenti o di ridotte dimensioni, indicativi di immunodeficienza cellulare; anomalie cutanee, quali alopecia, eczema, piodermiti o telangiectasia; evidenza di malattia ematologica, come pallore, petecchie, ittero o ulcere orali e linfoadenopatia generalizzata con splenomegalia, che può suggerire una malattia da HIV.
La linfocitopenia associata a stress, somministrazione di corticosteroidi, chemioterapia o irradiazione è, di solito, reversibile quando è rimosso l'agente o il fattore scatenante. L'efficace trattamento della malattia di base associata alla linfocitopenia (p. es. , malattia infettiva, infiammatoria e tumorale maligna) di solito determina un aumento della conta dei GB. Il trattamento ottimale dei pazienti con malattia da HIV richiede l'identificazione e il trattamento delle infezioni attive, profilassi primaria e secondaria per prevenire infezioni opportunistiche e il trattamento dell'infezione primaria da HIV per rallentare il deterioramento immunologico.
EMOSTASI L’emostasi normale è il processo attraverso il quale è mantenuta la fluidità del sangue nel sistema vascolare ed è consentita, quando e dove necessario (es. ferite, rotture dei vasi sanguigni), la rapida formazione di un solido tappo per tamponare l’emorragia. In condizioni di normalità c'è un continuo equilibrio fra fattori preposti a favorire la coagulazione locale del sangue ed i fattori che la controbilanciano (fibrinolisi). La salvaguardia dell'integrità della circolazione sanguigna è regolata da un complesso sistema di processi fisiologici multifattoriali che coinvolgono: piastrine vasi sanguigni (parete vascolare) fattori plasmatici solubili del sistema della coagulazione
Se per situazioni congenite o acquisite si verifica una alterazione di questi equilibri si possono determinare: Condizioni di ipercoagulabilità (trombosi) Fuoriuscita di sangue dai vasi sanguigni (emorragie)
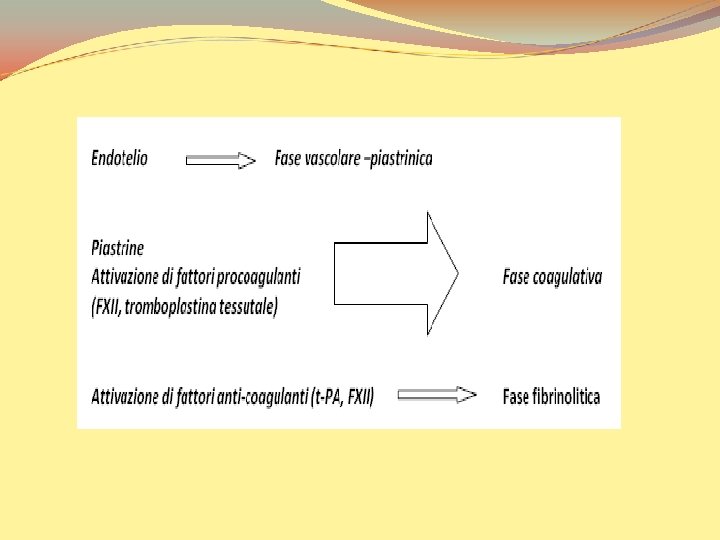
Dal punto di vista temporale il meccanismo dell’emostasi si articola in: 1. Breve periodo di VASOCOSTRIZIONE dovuto a meccanismi neurogeni riflessi e forse accentuato da fattori umorali come l’endotelina, un potente vasocostrittore di origine endoteliale. Tale contrazione è finalizzata alla riduzione tempestiva ma momentanea della perdita di sangue
2. La lesione endoteliale espone il tessuto connettivo subendoteliale altamente trombogenico, al quale le piastrine aderiscono iniziando la cosiddetta attivazione. Le piastrine attivate cambiano forma e liberano ADP, trombossano A 2 e serotonina. Sono così reclutate altre piastrine, che si aggregano alle prime e formano il tappo piastrinico: ciò avviene entro pochi minuti dalla lesione. E’ l’EMOSTASI PRIMARIA
3. Il rilascio di fattori tissutali nella sede della lesione attiva la sequenza della coagulazione del plasma, che culmina nella formazione della trombina (dalla protrombina). La trombina converte il fibrinogeno in fibrina, stimolando un ulteriore reclutamento di piastrine. Tutto ciò richiede tempo: è l’EMOSTASI SECONDARIA, SECONDARIA che porta alla formazione di una massa solida di fibrina e piastrine (tappo emostatico) a tamponare stabilmente l’emorragia. La funzione principale di tutta questa serie di eventi è quella di fornire una risposta rapida e localizzata che consenta la formazione di un tappo emostatico efficiente in grado di riparare la lesione vasale.
I meccanismi di regolazione dell’emostasi coinvolgono quindi: componenti cellulari e umorali del sangue circolante componenti cellulari: piastrine e leucociti componenti umorali: fibrinolisi fattori plasmatici della coagulazione e della sistema delle chinine sistema del complemento inibitori componenti dell’endotelio vasale (cellule endoteliali) reazioni vasomotorie che influenzano l’emodinamica: vasocostrizione e vasodilatazione determinate dalle varie sostanze secrete dall’endotelio vasale
La fase vascolare-piastrinica A seguito di una lesione vasale e della conseguente soluzione di continuità nelle strutture dell’endotelio, si verifica una immediata vasocostrizione riflessa accompagnata dal rallentamento del flusso ematico. Segue una immediata adesione delle piastrine all’endotelio attivata dalla esposizione delle fibrille di collagene della membrana basale dello strato subendoteliale del vaso (mediata da ADP e dal von Willebrand Factor, v. WF) L’adesione piastrinica provoca la “metamorfosi delle piastrine” le quali liberano così sostanze in grado di regolare la vasocostrizione/vasodilatazione del vaso (serotonina, fosfolipidi), sostanze che facilitano l’aggregazione piastrinica ( -TG), sostanze che regolano il complesso inibitore ATIII/Eparina (PF 4) o che attivano la cascata coagulatoria (fattore piastrinico P 3) Segue l’aggregazione delle piastrine conseguente formazione del cosiddetto “trombo bianco” che rappresenta il primo tappo emostatico
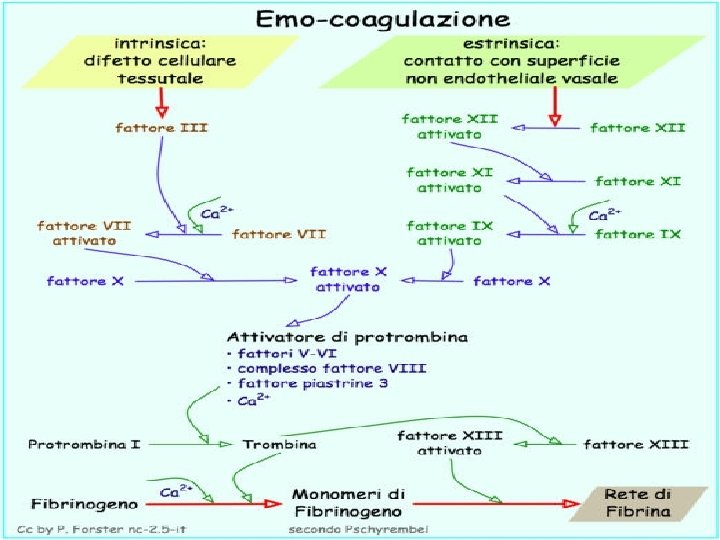
La fase coagulatoria La lesione del vaso provoca l’attivazione a cascata dei processi che portano alla formazione di un coagulo in grado di riparare più efficacemente e stabilmente la lesione del vaso stesso. L’esposizione di molecole cariche negativamente da parte del’endotelio danneggiato induce l’attivazione per contatto della via intrinseca della coagulazione (FXII); la liberazione della tromboplastina tessutale da parte dei tessuti danneggiati attiva la via estrinseca (FVII). Per entrambi i processi è fondamentale la liberazione di fosfolipidi da parte delle piastrine (P 3). Sia l’attivazione della via intrinseca sia di quella estrinseca provocano l’attivazione a cascata di numerosi fattori che inducono alla fine la trasformazione del fibrinogeno, la
La fibrinoformazione Il punto finale del processo coagulativo è la produzione del coagulo di fibrina. La Trombina distacca due frammenti polipeptidici dalle catene Aa e Bb del fibrinogeno (detti FPA e FPB). Si ottengono così dei monomeri di fibrina (FM) che tendono a polimerizzare dando origine a molecole di fibrina solubile che, per azione del FXIII, si stabilizza in fibrina insolubile. Strettamente connesso al sistema emocoagulativo è il sistema fibrinolitico che, ad opera della plasmina, digerisce fibrina e fibrinogeno dando origine ai loro prodotti di degradazione D-D e FDP.
Il tempo di tromboplastina parziale (PTT): è sensibile a deficit di circa il 30 -40% di tutti i fattori della coagulazione eccetto che per i fattori VII e XIII. Con rare eccezioni, un risultato normale esclude l’emofilia. L’eparina prolunga il PTT e quest’ultimo viene utilizzato spesso per il monitoraggio della terapia eparinica. Un tempo prolungato può anche essere causato da un deficit di uno o più fattori della coagulazione o dalla presenza di un inibitore di un fattore della coagulazione (p. es. , un anticoagulante che inibisce il fattore VIII) o di un inibitore del fosfolipide procoagulante. Nel test del tempo di protrombina (PT): Il test individua i casi in cui siano presenti anomalie dei fattori V, VII e X, della protrombina e del fibrinogeno; il PT normale varia da 10 e 12 sec, in rapporto al particolare reagente impiegato contenente fattore tissutale e di altri dettagli tecnici. Un PT più lungo di 2 s rispetto al valore normale, di controllo del laboratorio, deve essere considerato anormale e ne va indagato il motivo. Il PT è un esame utile per lo screening di disturbi della coagulazione in varie condizioni acquisite (p. es. , carenza di vitamina K, epatopatie, CID). Il PT viene anche impiegato per controllare la terapia con anticoagulanti cumarinici. Il range terapeutico del PT dipende dalla tromboplastina utilizzata in ogni laboratorio. Il rapporto internazionale normalizzato (INR, valore normale 0, 9 -1, 1) è stato introdotto dalla WHO per standardizzare internazionalmente il controllo della terapia anticoagulante. L’INR è il rapporto del PT del paziente rispetto al controllo elevato all’indice di sensibilità internazionale (ISI):
Disordini dell’emostasi e della coagulazione Malattie caratterizzate da una tendenza al sanguinamento.
Disordini della coagulazione ereditari Emofilia Forme comuni di disordini emorragici ereditari causate da deficit di fattore VIII, IX o XI della coagulazione. L’emofilia A (deficienza del fattore VIII), che colpisce circa l’ 80% degli emofiliaci e l’emofilia B (deficienza del fattore IX) hanno identiche manifestazioni cliniche, anomalie dei test di screening, così come identica trasmissione genetica, legata al cromosoma X. Il dosaggio degli specifici fattori è necessario per distinguere le due forme. Poiché i geni dei fattori VIII e IX sono localizzati sul cromosoma X, l’emofilia colpisce pressoché esclusivamente i maschi. Le figlie degli emofilici saranno portatrici obbligate, mentre tutti i figli maschi saranno normali. Ciascun figlio maschio di una portatrice avrà il 50% delle possibilità di essere emofilico e ciascuna figlia ha il 50% delle possibilità di essere portatrice.
Malattie coagulative ereditarie rare Esistono altre condizioni di deficit ereditario di fattori della coagulazione. La maggior parte è costituita da rari stati di tipo autosomico recessivo che causano la malattia solo nella condizione omozigote. Il deficit di Fattore XI è inusuale nella popolazione generale ma comune nei discendenti degli Ebrei europei (frequenza del gene, 5 -9%). Un disordine emorragico caratterizzato da sanguinamento da lesione (trauma o chirurgia) si verifica negli omozigoti e nei doppi eterozigoti e occasionalmente negli eterozigoti. Un altro importante disordine deriva da una deficienza di α 2 antiplasmina, il principale inibitore fisiologico della plasmina. Il dosaggio specifico dell’α 2 -antiplasmina mostrerà valori dell’ 1 -3% del range normale. La profilassi con acido δ-aminocaproico o acido tranexamico correggerà la tendenza emorragica. Un eterozigote, con un tasso di α 2 -antiplasmina nel 30 -40% del valore normale, può presentare un eccessivo sanguinamento chirurgico, se si manifesta un grado inusuale di fibrinolisi.
Coagulopatie acquisite Disordini della coagulazione correlati a malattie epatiche Malattie epatiche possono disturbare l’emostasi danneggiando la sintesi dei fattori coagulativi, aumentando la fibrinolisi o causando trombocitopenia. Nei pazienti con epatite fulminante o con fegato grasso acuto in corso di gravidanza, l’emostasi è disturbata a causa della ridotta produzione e del consumo di fattori della coagulazione nei trombi intravascolari.
Coagulazione intravascolare disseminata (CID, coagulopatia da consumo) Anomala generazione di fibrina nel sangue circolante. La coagulazione intravascolare disseminata (CID) consegue generalmente all’ingresso o alla formazione nel circolo ematico di materiale con attività di fattore tissutale (TFA), che dà inizio alla coagulazione. Di solito la CID si verifica in una delle seguenti quattro condizioni cliniche: 1. In corso di complicanze ostetriche, abruptio placentae, nell’aborto terapeutico indotto con soluzione fisiologica, nella sindrome da ritenzione del feto morto e nella fase iniziale dell’embolia amniotica. Materiale uterino con attività di fattore tissutale ha accesso al circolo materno. 2. In corso di infezioni, soprattutto da gram -, la cui endotossina causa generazione di attività di fattore tissutale sulla membrana citoplasmatica dei monociti e delle cellule endoteliali. 3. In presenza di neoplasia: particolarmente in caso di adenocarcinomi secernenti mucina del pancreas e della prostata e della leucemia promielocitica acuta. 4. Shock da qualsiasi causa, probabilmente a causa della generazione di attività di fattore tissutale su monociti e cellule endoteliali.
Disordini della coagulazione da anticoagulanti circolanti Gli anticoagulanti circolanti sono sostanze endogene che inibiscono la coagulazione. Tali sostanze sono generalmente anticorpi che neutralizzano l’attività di uno dei fattori della coagulazione (p. es. , un anticorpo contro il fattore VIII o il fattore V) o l’attività del fosfolipide procoagulante. Raramente gli anticoagulanti circolanti sono glicosaminoglicani con attività anticoagulante eparino-simile derivante dalla loro capacità di aumentare l’attività dell’antitrombina III. Tali anticoagulanti eparinosimili si rilevano soprattutto in pazienti con mieloma multiplo o altre affezioni neoplastiche maligne ematologiche.
Disordini trombotici Malattie caratterizzate da formazione di un trombo che ostruisce il flusso ematico vascolare localmente o che si stacca ed embolizza per interrompere il flusso ematico a distanza (tromboembolismo).
I trombi sono masse meccaniche si formano all’interno del sistema cardiovascolare o sulla superficie endovascolare o protesica denudata. Sono composti di fibrina insolubile, piastrine depositate, GB che si accumulano e GR intrappolati in assetti variabili dipendenti dal flusso. La formazione del trombo è un processo multifattoriale che coinvolge molti fattori genetici e ambientali mutualmente interattivi. Dei pazienti con trombosi venosa profonda dimostrata venograficamente, il 25 -50% presenta un fattore genetico predisponente. Un meccanismo anticoagulante geneticamente difettoso (p. es. , resistenza alla proteina C attivata, iperomocisteinemia, deficit di proteina C, deficit di proteina S, deficit di antitrombina III, insufficiente fibrinolisi), quando combinato con uno stimolo trombotico (p. es. , chirurgia, gravidanza, uso di contraccettivi orali, anticorpi antifosfolipidi), è sufficiente per sviluppare un tromboembolismo venoso. La terapia antitrombotica include l’uso di farmaci trombolitici, farmaci antipiastrinici e anticoagulanti. Le misure meccaniche per ripristinare la piena funzionalità vascolare includono un catetere a palloncino e l’embolectomia chirurgica.
Resistenza del fattore V alla proteina C attivata Iperomocisteinemia Deficit di proteina C Deficit di proteina S Carenza di antitrombina Ridotta fibrinolisi Sindrome da fosfolipidi Aterosclerosi Altri fattori predisponenti: Stasi Danno tissutale da trauma e da atto chirurgico Neoplasie Processi infiammatori cronici Apparati protesici cardiovascolari farmaci contraccettivi orali
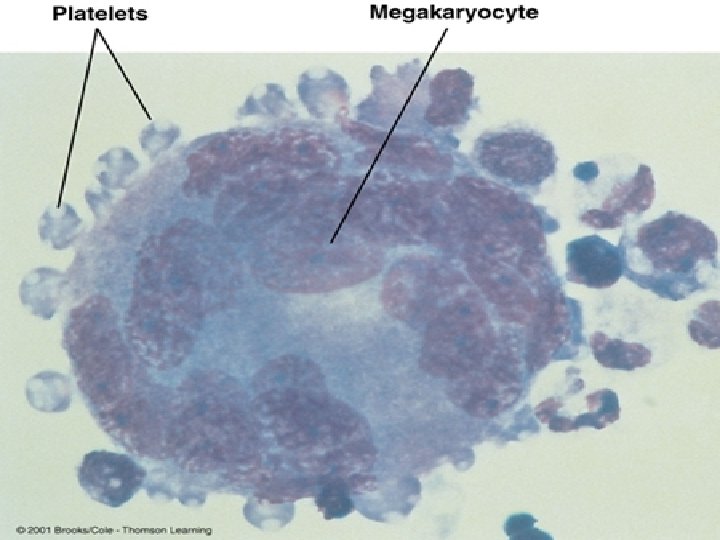
Piastrine Le piastrine non sono vere cellule. Sono prodotte per gemmazione dai megacariociti. Sono dischetti di piccole dimensioni (circa 3 µm). La principale funzione delle piastrine, o trombociti, è di fermare la perdita di sangue nelle ferite (emostasi). Anche se appaiono di forma tondeggiante, le piastrine non sono propriamente delle cellule. Negli strisci colorati con il giemsa, hanno un colore porpora intenso. Il loro diametro è di circa 2 -3 µm, quindi sono assai più piccole degli eritrociti. Valori normali: da 140. 000 a 440. 000 /mmc
Patologie piastriniche possono causare una difettosa formazione dei coaguli emostatici e sanguinamenti: Riduzione del numero delle piastrine (trombocitopenia) Ridotta funzione piastrinica, nonostante un numero normale (disfunzione piastrinica).
Trombocitemia Una quantità di piastrine sotto il normale range di 140000 -440000/ml.
Trombocitemia essenziale (Trombocitemia primitiva) Malattia caratterizzata da un aumento delle piastrine, da iperplasia megacariocitica e da diatesi emorragica o trombotica. La trombocitemia primitiva è un’anomalia clonale di una cellula staminale emopoietica multipotente. Si manifesta di solito tra i 50 e i 70 anni e colpisce in ugual misura sia gli uomini che le donne. Il notevole aumento della conta delle piastrine risulta da un aumento della loro produzione. La sopravvivenza delle piastrine è generalmente normale, ma può anche risultare ridotta a causa del sequestro splenico.
Trombocitemia secondaria La trombocitemia secondaria è un processo reattivo. La conta piastrinica è < 1 - 106/µl e la causa può essere ovvia sulla base dell’anamnesi o dell’esame clinico I test di funzione piastrinica di solito risultano normali. Tuttavia, in malattie mieloproliferative, un’anomala aggregabilità piastrinica si rileva in circa il 50% dei pazienti. La terapia della trombocitemia secondaria è rappresentata da quella della patologia di base. Con terapia appropriata, la conta piastrinica generalmente si normalizza.
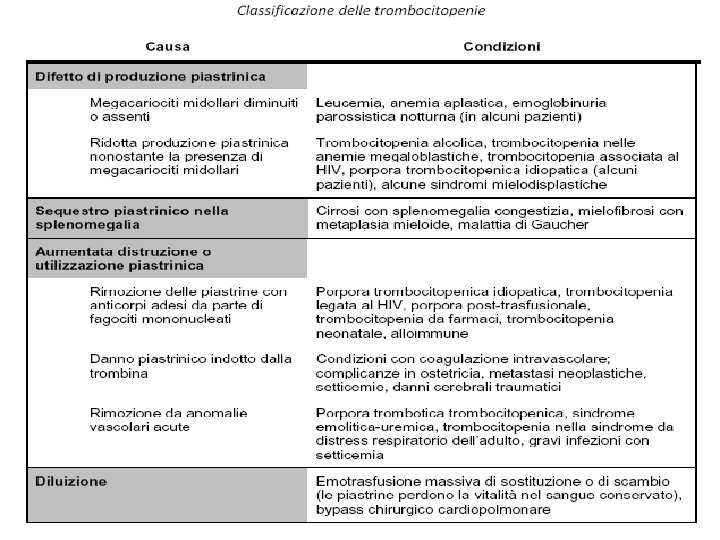
Trombocitopenia Una quantità di piastrine sotto il normale range di 140000 -440000/ml.
Una trombocitopenia può essere espressione di: una ridotta produzione piastrinica sequestro splenico aumentata distruzione utilizzazione o diluizione piastrinica. Indipendentemente dalla causa, una grave trombocitopenia provoca un quadro emorragico tipico: petecchie multiple cutanee, evidenziabili in particolare verso l’estremità inferiore delle gambe e di piccole ecchimosi sparse in seguito a traumi di lieve entità sanguinamenti delle mucose (epistassi, emorragie del tratto GI, GU, sanguinamento vaginale) e sanguinamento eccessivo dopo interventi chirurgici.
Disfunzioni piastriniche In alcune malattie la conta piastrinica può risultare normale, ma le piastrine possono essere incapaci di formare normalmente coaguli emostatici, prolungando il tempo di sanguinamento. L’alterata funzione piastrinica può conseguire a un difetto intrinseco piastrinico o a un fattore estrinseco che alteri la funzione di piastrine per il resto normali. I difetti possono essere ereditari o acquisiti. I test delle fasi della coagulazione dell’emostasi (p. es. , tempo di tromboplastina parziale e tempo di protrombina [PT]) sono normali nella maggior parte dei casi.
Difetti ereditari della funzione piastrinica Quando dall’anamnesi dell’infanzia di un paziente risulta una facilità alle ecchimosi e alle emorragie dopo estrazioni dentarie, tonsillectomia o altri interventi chirurgici, il rilievo di una normale conta piastrinica con un prolungato tempo di sanguinamento suggerisce un difetto ereditario della funzione piastrinica. La causa può essere la malattia di Von Willebrand, la più comune malattia emorragica ereditaria, o un difetto intrinseco piastrinico ereditario, che è molto meno comune.
Difetti piastrinici acquisiti Molti disordini clinici (p. es. , malattie mieloproliferative e disordini mielodisplastici, uremia, macroglobulinemia e mieloma multiplo, cirrosi e LES) possono anche influenzare la funzione piastrinica. L’aspirina, che allunga di poco il tempo di sanguinamento in molte persone sane, lo può allungare marcatamente in pazienti con una sottostante disfunzione piastrinica o con una grave alterazione della coagulazione. Le piastrine possono presentare una disfunzione, portando a un allungamento del tempo di sanguinamento, come si verifica quando il sangue passa attraverso un ossigenatore a pompa durante interventi cardiochirurgici di bypass. I pazienti con uremia causata da insufficienza renale cronica possono presentare un lungo tempo di sanguinamento per motivi sconosciuti.