Human Genetic Disorders Genetic Linkage and Counseling Genetic



 of the")




, or sickle-cell anaemia, is an autosomal recessive genetic blood disorder, characterized")
, the polymerisation problems are")
 is synthesized in a complex series of steps. The heme part is")

 are a group of inherited blood disorders caused by reduced or absent")
 - In adults Hemoglobin F is restricted to a")



 is an inherited condition in which numerous polyps form mainly")
 gene is located on the long (q) arm")

, is a type of inherited")
 is a rare genetic disorder in which seven")

 is a neurodegenerative genetic disorder that affects muscle coordination and leads")



, also known in medical literature as Alzheimer disease, is the most")


 and the Golgi apparatus (1). Following")

 is among the newest and most sophisticated of")

 is a form of prenatal diagnosis to determine chromosomal or")

 is a method used to genotype human embryos for known")
 has led to this procedure, allowing parents to weed out")

 officially defines genetic counseling as the process")


- Slides: 46
Human Genetic Disorders, Genetic Linkage and Counseling
Genetic Linkage
Genetic linkage is the tendency of certain loci or alleles to be inherited together. Alleles that are located on the same chromosome tend to stay together during meiosis and are thus genetically linked. Chromosomal recombination during prophase I of Meiosis prevents alleles originally located on the same chromosome from being permanently linked.
Chromosome mapping by counting recombinant phenotypes produces a genetic map (linkage map) of the chromosome. %age of recombination (recombination frequency) is used as a unit, called centimorgan, after Thomas Morgan who described it: If only 3. 0% of the gametes contain a recombinant chromosome, the two genes are said to be 3 c. M apart on the chromosome. . . But all the genes on the chromosome are incorporated in a single molecule of DNA. Genes are simply portions of the molecule (open reading frames or ORFs) encoding products that create the observed trait (phenotype). The rapid progress in DNA sequencing has produced complete genomes for hundreds of microbes and several eukaryotes. As a very rough rule of thumb, 1 c. M on a chromosome encompasses 1 megabase (1 Mb = 106 bp) of DNA. But for the reasons mentioned above, this relationship is only approximate. Although the genetic maps of human females average 90% longer than the same maps in males, their chromosomes contain the same number of base pairs. So their physical maps are identical. http: //users. rcn. com/jkimball. ma. ultranet/Biology. Pages/L/Linkage. html
A genetic map of the genes affecting adult height. Genetic linkage analysis was used for locating genes affecting stature. This method utilizes genetic markers known to show variation between individuals. The markers are evenly distributed across the entire genome and they are determined from DNA samples. By using these markers it is possible to determine, by means of the linkage analysis, the chromosomal regions that are similar in family members showing a strong correlation in height. As a result, scientists can estimate the statistical probability of the region containing genes associated with adult stature. Statistical significance for the detected linkages must then be determined through computer simulation. When populations share genetic background and environmental factors, average height is frequently characteristic within the group. Exceptional height variation (around 20% deviation from average) within such a population is usually due to gigantism or dwarfism; which are medical conditions due to specific genes or to endocrine abnormalities. In regions of extreme poverty or prolonged warfare, environmental factors like malnutrition during childhood or adolescence may account for marked reductions in adult stature even without the presence of any of these medical conditions. Human height is 60%– 80% heritable, according to several twin studies and has been considered polygenic since the Mendelian-biometrician debate a hundred years ago. The only gene known to have an influence on human height is HMGA 2. People who carry two copies of the "tall" allele of the HMGA 2 gene are up to 1 cm taller than those who carry two copies of the "short" allele. A genome-wide association (GWA) study of more than 180, 000 individuals has identified hundreds of genetic variants in at least 180 loci associated with adult human height
Human Genetic Disorders
Amelogenesis imperfecta presents with abnormal formation of the enamel or external layer of teeth. Enamel is composed mostly of mineral, that is formed and regulated by the proteins in it. Amelogenesis imperfecta is due to the malfunction of the proteins in the enamel: ameloblastin, enamelin, tuftelin and amelogenin. People afflicted with amelogenesis imperfecta have teeth with abnormal color: yellow, brown or grey. The teeth have a higher risk for dental cavities and are hypersensitive to temperature changes. This disorder can afflict any number of teeth. Up to date, mutations in the AMELX, ENAM, MMP 20, and KLK-4 genes have been found to cause amelogenesis imperfecta (non-syndromic form). The AMELX, ENAM, KLK-4 and MMP 20 genes are essential for normal tooth development. These proteins are involved in the formation of enamel, which is a hard, calcium-rich material that forms the protective outer layer of each tooth. Amelogenesis imperfecta that results from mutations in the ENAM gene is inherited in an autosomal dominant pattern. This type of inheritance means one copy of the altered gene in each cell is sufficient to cause the disorder. Amelogenesis imperfecta is also inherited in an autosomal recessive pattern; this form of the disorder can result from mutations in the ENAM or MMP 20 gene. About 5% of amelogenesis imperfecta cases are caused by mutations in the AMELX gene and are inherited in an X -linked pattern. In most cases, males with an X-linked form of this condition experience more severe dental abnormalities than affected females.
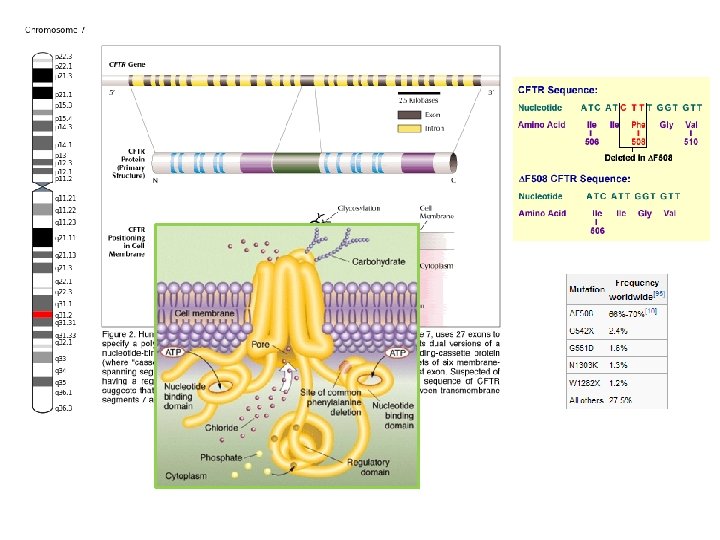
Cystic fibrosis is an autosomal recessive genetic disorder affecting most critically the lungs, and also the pancreas, liver, and intestine. It is characterized by abnormal transport of chloride and sodium across an epithelium, leading to thick, viscous secretions. The name cystic fibrosis refers to the characteristic scarring (fibrosis) and cyst formation within the pancreas, first recognized in the 1930 s. Difficulty breathing is the most serious symptom and results from frequent lung infections that are treated with antibiotics and other medications. CF is caused by a mutation in the gene for the protein cystic fibrosis transmembrane conductance regulator (CFTR). This protein is required to regulate the components of sweat, digestive juices, and mucus. CFTR regulates the movement of chloride and sodium ions across epithelial membranes, such as the alveolar epithelia located in the lungs. Although most people without CF have two working copies of the CFTR gene, only one is needed to prevent cystic fibrosis due to the disorder's recessive nature. CF develops when neither gene works normally (as a result of mutation) and therefore has autosomal recessive inheritance. (pleiotropic effects of a single gene mutation)
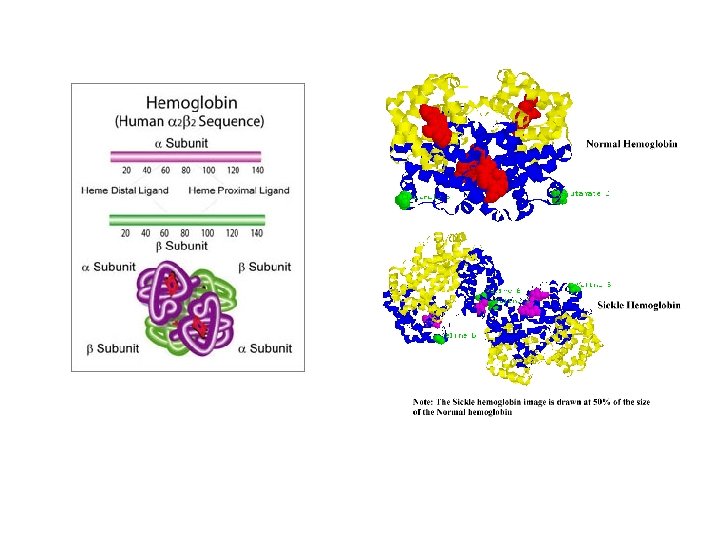
Sickle-cell disease (SCD), or sickle-cell anaemia, is an autosomal recessive genetic blood disorder, characterized by red blood cells that assume an abnormal, rigid, sickle shape. The sickling occurs because of a mutation in the hemoglobin gene. Life expectancy is shortened. In 1994, in the US, the average life expectancy of persons with this condition was estimated to be 42 years in males and 48 years in females. Sickle-cell disease, usually presenting in childhood, occurs more commonly in people from parts of tropical and sub-tropical regions where malaria is or was common. One-third of all indigenous inhabitants of Sub-Saharan Africa carry the gene, because in areas where malaria is common, there is a fitness benefit in carrying only a single sickle-cell gene (heterozygous for the mutation). Those with only one of the two alleles of the sickle-cell disease, while not totally resistant, are more tolerant to the infection and thus show less severe symptoms when infected (hence the mutation does not get eliminated from the gene pool)
In people heterozygous for Hgb. S (carriers of sickling haemoglobin), the polymerisation problems are minor, because the normal allele is able to produce over 50% of the haemoglobin. Carriers have symptoms only if they are deprived of oxygen (for example, while climbing a mountain) or while severely dehydrated. In people homozygous for Hgb. S, the presence of long-chain polymers of Hb. S distort the shape of the red blood cell from a smooth doughnut-like shape to ragged and full of spikes, making it fragile and susceptible to breaking within capillaries. The sickle-cell disease occurs when the seventh amino acid (if the initial methionine is counted), glutamic acid, is replaced by valine to change its structure and function. Valine is hydrophobic, causing the haemoglobin to collapse in on itself occasionally. The structure is not changed otherwise. When enough haemoglobin collapses in on itself the red blood cells become sickle-shaped.
Hemoglobin (Hb) is synthesized in a complex series of steps. The heme part is synthesized in a series of steps in the mitochondria and the cytosol of immature red blood cells, while the globin protein parts are synthesized by ribosomes in the cytosol. Production of Hb continues in the cell throughout its early development from the proerythroblast to the reticulocyte in the bone marrow. At this point, the nucleus is lost in mammalian red blood cells, but not in birds and many other species. Even after the loss of the nucleus in mammals, residual ribosomal RNA allows further synthesis of Hb until the reticulocyte loses its RNA soon after entering the vasculature (this hemoglobin-synthetic RNA in fact gives the reticulocyte its reticulated appearance and name). Glycine is the precursor of porphyrins. Hemoglobin A (α 2β 2) is the most common form, with a normal amount over 95% (in normal individuals)
The α thalassemias involve the genes HBA 1 and HBA 2, inherited in a Mendelian recessive fashion. There are two gene loci and so four alleles. It is also connected to the deletion of the 16 p chromosome. α Thalassemias result in decreased alpha-globin production, therefore fewer alpha-globin chains are produced, resulting in an excess of β chains in adults and excess γ chains in newborns. The excess β chains form unstable tetramers (called Hemoglobin H or Hb. H of 4 beta chains), which have abnormal oxygen dissociation curves. Hemoglobin H (β 4) - A variant form of hemoglobin, formed by a tetramer of β chains, which may be present in variants of α thalassemia. Hemoglobin Barts (γ 4) - A variant form of hemoglobin, formed by a tetramer of γ chains, which may be present in variants of α thalassemia.
Beta-thalassemias (β-thalassemias) are a group of inherited blood disorders caused by reduced or absent synthesis of the beta chains of hemoglobin resulting in variable phenotypes ranging from severe anemia to clinically asymptomatic individuals. The total annual incidence of symptomatic individuals is estimated at 1 in 100, 000 throughout the world. Three main forms have been described: thalassemia major, thalassemia intermedia and thalassemia minor. Individuals with beta thalassemia major usually present within the first two years of life with severe anemia, poor growth, and skeletal abnormalities during infancy. Affected children will require regular lifelong blood transfusions. Beta thalassemia intermedia is less severe than beta thalassemia major and may require episodic blood transfusions. Transfusion-dependent patients will develop iron overload and require chelation therapy to remove the excess iron. Bone marrow transplants can be curative for some children with beta thalassemia major. Transmission is autosomal recessive; however, dominant mutations have also been reported.
Hemoglobin F (α 2γ 2) - In adults Hemoglobin F is restricted to a limited population of red cells called F-cells. However, the level of Hb F can be elevated in persons with sickle-cell disease and betathalassemia. Hemoglobin C (α 2βC 2) - Another variant due to a variation in the β-chain gene. This variant causes a mild chronic hemolytic anemia. Hemoglobin E (α 2βE 2) - Another variant due to a variation in the β-chain gene. This variant causes a mild chronic hemolytic anemia. Hemoglobin S (α 2βS 2) - A variant form of hemoglobin found in people with sickle cell disease. There is a variation in the β-chain gene, causing a change in the properties of hemoglobin, which results in sickling of red blood cells.
Knowing that HBA 1 and HBA 2 encodes alpha-globin and is associated with alpha thalassemia, whom is localized at the telomeric region chromosome 16 and is cluster together with regulatory element. All regulatory-element mutation ultimately alter expresion of HBA 1 and HBA 2. By doing an analysis of the entire coding region, detection of the mutation or deletion of genes is possible. Other testing such as Sequence analysis (to identify point mutation), Targeted Mutation analysis (involving deletion of alpha-globin or sequence variants) , and Deletion/duplication analysis is also possible.
Tay–Sachs disease is an autosomal recessive genetic disorder. In its most common variant (known as infantile Tay –Sachs disease), it causes a progressive deterioration of mental and physical abilities that commences around six months of age and usually results in death by the age of four. Tay–Sachs disease is caused by a genetic defect, normally inherited from both parents. The disease occurs when harmful quantities of cell membrane components known as gangliosides accumulate in the brain's nerve cells, eventually leading to the affected cells' premature death. Currently, there is no known cure or treatment. Cherry-red spot as seen in Tay Sachs disease: the fovea's center appears bright red because it is surrounded by a milky halo
Research in the late 20 th century demonstrated that Tay –Sachs disease is caused by a genetic mutation in the HEXA gene on (human) chromosome 15. The GM 2 gangliosidotic diseases are severe psycho-motor developmental disorders caused by the inability to properly degrade membrane associated gangliosides of the GM 2 family. Hexosaminidase is a dimer composed of 2 subunits, either the α subunit (encoded by the HEXA gene) and/or the β subunit (encoded by the HEXB gene). The degradation of GM 2 gangliosides takes place in the lysosomes. Failure to degrade these sphingolipids results in the lysosomes becoming engorged filling the cell, eventually choking off normal cellular functions. Because of the deposition of abnormal sphingolipids in the lysosomes the GM 2 gangliosidotic diseases are also referred to as lysosomal storage diseases. Because neural cell membranes are enriched in GM 2 gangliosides, the inability to degrade this class of sphingolipid results in neural cell death. HEXA mutations are rare and are most seen in genetically isolated populations. Tay–Sachs can occur from the inheritance of either two similar, or two unrelated, causative mutations in the HEXA gene.
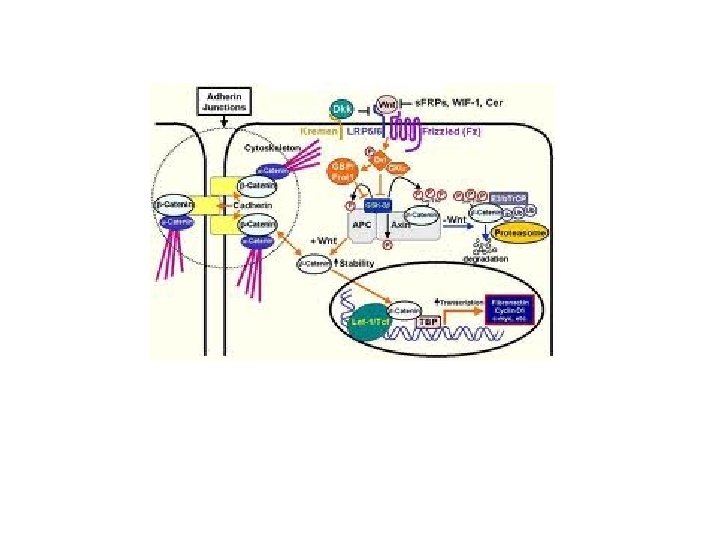
Familial adenomatous polyposis (FAP) is an inherited condition in which numerous polyps form mainly in the epithelium of the large intestine. While these polyps start out benign, malignant transformation into colon cancer occurs when not treated. Familial adenomatous polyposis can have different inheritance patterns and different genetic causes. When this condition results from mutations in the APC gene, it is inherited in an autosomal dominant pattern, which means one copy of the altered gene is sufficient to cause the disorder. (APC is a tumor suppressor gene. ) The incidence of malignancy in these cases approaches 100%. In most cases, an affected person has one parent with the condition. Mutations in the MUTYH gene are inherited in an autosomal recessive pattern, which means two copies of the gene must be altered for a person to be affected by the disorder. (MUTYH is a DNA glycosylase, functioning in DNA repair). Most often, the parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene.
The human APC (adenomatous polyposis coli) gene is located on the long (q) arm of chromosome 5 between positions 21 and 22. . . More than 800 mutations in the APC gene have been identified in families with classic and attenuated types of familial adenomatous polyposis. Most of these mutations cause the production of an APC protein that is abnormally short and nonfunctional. This short protein cannot suppress the cellular overgrowth that leads to the formation of polyps, which can become cancerous.
The flat surface of the colon is covered by an epithelium composed of four differentiated cell types (enterocytes, enteroendocrine, goblet and Paneth cells) that invaginates at regular intervals to form crypts (fig 1). The bottom of the crypts is occupied by a few stem cells that give rise to actively dividing precursor cells that populate the bottom two-thirds of the crypts. 1 Proliferation occurs under the influence of growth factors from the Wnt family that may be produced by the underlying stromal cells underneath the stem cell compartment or by the epithelial cells themselves. The precursors migrate upward in an ordered fashion, which is also controlled by Wnt factors, 2 and they stop proliferation when they reach the top third of the crypt, probably because they are too far from the Wnt source. Meanwhile, they continue their migration movement and colonise the surface of the colon. After about a week, epithelial cells undergo apoptosis and are shed in the lumen of the gut. http: //gut. bmj. com/content/56/3/417. extract
Lynch syndrome, often called hereditary nonpolyposis colorectal cancer (HNPCC), is a type of inherited cancer of the digestive tract (autosomal dominant), particularly the colon (large intestine) and rectum. People with Lynch syndrome have an increased risk of cancers of the stomach, small intestine, liver, gallbladder ducts, upper urinary tract, brain, skin, and prostate. Women with this disorder also have a high risk of cancer of the endometrium (lining of the uterus) and ovaries. Even though the disorder was originally described as not involving noncancerous (benign) growths (polyps) in the colon, people with Lynch syndrome may occasionally have colon polyps. In individuals with this disorder, colon polyps occur at an earlier age than in the general population. Although the polyps do not occur in greater numbers than in the general population, they are more likely to become cancerous. Variations in the MLH 1, MSH 2, MSH 6, and PMS 2 genes increase the risk of developing Lynch syndrome. All of these genes are involved in the repair of mistakes made when DNA is copied (DNA replication) in preparation for cell division. Mutations in any of these genes prevent the proper repair of DNA replication mistakes. As the abnormal cells continue to divide, the accumulated mistakes can lead to uncontrolled cell growth and possibly cancer.
Prader–Willi syndrome ( ; abbreviated PWS) is a rare genetic disorder in which seven genes (or some subset thereof) on chromosome 15 (q 11– 13) are deleted or unexpressed (chromosome 15 q partial deletion) on the paternal chromosome. It was first described in 1956 by Andrea Prader (1919– 2001), Heinrich Willi (1900– 1971), Alexis Labhart (1916), Andrew Ziegler, and Guido Fanconi of Switzerland. Characteristic of PWS is "low muscle tone, short stature, incomplete sexual development, cognitive disabilities, problem behaviors, and a chronic feeling of hunger that can lead to excessive eating and lifethreatening obesity. ” The incidence of PWS is between 1 in 25, 000 and 1 in 10, 000 live births. The paternal origin of the genetic material that is affected in the syndrome is important because the particular region of chromosome 15 involved is subject to parent of origin imprinting, meaning that for a number of genes in this region only one copy of the gene is expressed while the other is silenced through imprinting. For the genes affected in PWS, it is the paternal copy that is usually expressed, while the maternal copy is silenced.
PWS is caused by the deletion of the paternal copies of the imprinted SNRPN and necdin genes along with clusters of sno. RNAs. Due to imprinting, the maternally inherited copies of these genes are virtually silent, only the paternal copies of the genes are expressed. PWS results from the loss of paternal copies of this region. . . Prader–Willi syndrome has no cure; however, several treatments are in place to lessen the condition's symptoms. During infancy, subjects should undergo therapies to improve muscle tone. Speech and occupational therapy are also indicated. During the school years, children benefit from a highly structured learning environment as well as extra help. The largest problem associated with the syndrome is severe obesity.
Huntington's disease (HD) is a neurodegenerative genetic disorder that affects muscle coordination and leads to cognitive decline and psychiatric problems. It typically becomes noticeable in mid-adult life. HD is the most common genetic cause of abnormal involuntary writhing movements. The disease is caused by an autosomal dominant mutation in either of an individual's two copies of a gene called Huntingtin, which means any child of an affected parent has a 50% risk of inheriting the disease. Physical symptoms of Huntington's disease can begin at any age from infancy to old age, but usually begin between 35 and 44 years of age. The Huntingtin gene provides the genetic information for a protein that is also called "huntingtin". Expansion of a CAG triplet repeat stretch within the Huntingtin gene results in a different (mutant) form of the protein, which gradually damages cells in the brain, through mechanisms that are not fully understood.
Htt is expressed in all mammalian cells. The highest concentrations are found in the brain and testes, with moderate amounts in the liver, heart, and lungs. The function of Htt in humans is unclear. It interacts with proteins which are involved in transcription, cell signaling and intracellular transporting. In animals genetically modified to exhibit HD, Htt was found to be important for embryonic development, as its absence is related to embryonic death. It also acts as an anti-apoptotic agent preventing programmed cell death and controls the production of brain-derived neurotrophic factor, a protein which protects neurons and regulates their creation during neurogenesis. Htt also facilitates vesicular transport and synaptic transmission and controls neuronal gene transcription. CAG repeat extension (poly. Q diseases; CAG codes for Glutamine, Q)
http: //www. pnas. org/content/97/24/12957/F 1. expansion. html
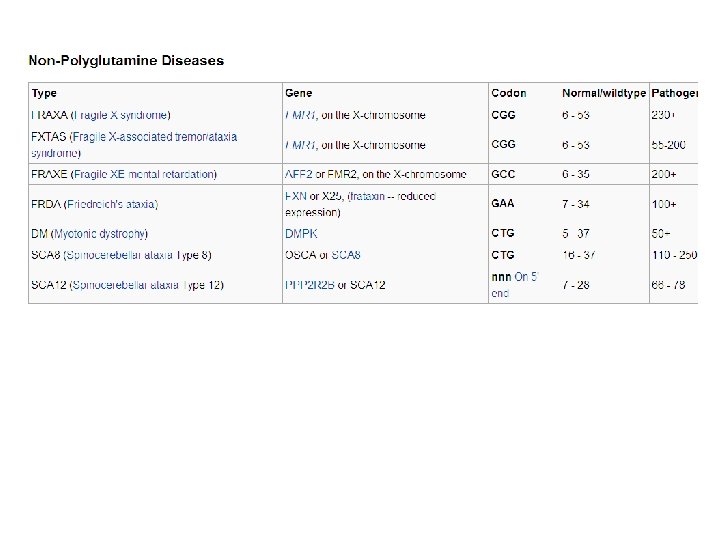
What are polyglutamine diseases? Trinucleotide repeat disorders are a set of genetic disorders caused by trinucleotide repeat expansion, a kind of mutation where trinucleotide repeats in certain genes exceed the normal, stable, threshold, which differs per gene. (If there is an expansion of CAG, which codes for Gln, then it is called a polyglutamine disease; Fragile X syndrome is caused by an expansion of CGG repeat, Huntington's disease by CAG repeat, Friedreich's ataxia by GAA repeat and Myotonic dystrophy by CTG repeat. ).
Alzheimer's disease (AD), also known in medical literature as Alzheimer disease, is the most common form of dementia. There is no cure for the disease, which worsens as it progresses, and eventually leads to death. It was first described by German psychiatrist and neuropathologist Alois Alzheimer in 1906 and was named after him. The vast majority of cases of Alzheimer's disease are sporadic, meaning that they are not genetically inherited although some genes may act as risk factors. On the other hand, around 0. 1% of the cases are familial forms of autosomal dominant (not sex-linked) inheritance, which usually have an onset before age 65. This form of the disease is known as Early onset familial Alzheimer's disease.
http: //hmg. oxfordjournals. org/content/19/R 1/R 4/F 3. expansion. html
Schematic showing the role of cholesterol-rich lipid rafts in segregating the non-amyloidogenic proteolytic processing of APP by the α-secretase (B) from the amyloidogenic processing by β- and γ-secretases http: //www. nature. com/nrn/journal/v 8/n 7/fig_tab/nrn 2168_F 3. html This neurodegenerative disorder is characterized by the progressive deposition of the 4 k. Da amyloid-β (Aβ) peptide in extracellular senile plaques in the brain. Aβ is derived by proteolytic cleavage of the amyloid precursor protein (APP), a Type I integral membrane protein. Cleavage of APP at the Nterminus of the Aβ by β-secretase (BACE 1) and at the C-terminus by the γ-secretase complex (presenilins, nicastrin, Aph 1 and Pen 2) constitutes the amyloidogenic pathway for processing of APP.
APP is synthesized by the endoplasmatic reticulum (ER) and the Golgi apparatus (1). Following the amyloidogenic pathway in neurons, APP is cleaved by β-secretase (BACE 1) and γ-secretase (PSEN) to generate Aβ peptides and the amyloid intracellular domain [AICD] (2), which influences the transcription of several genes (3). In the APP retromer recycling pathway (1), APP is redirected to endosomes by SORL 1. PICALM has a presumed role in APP endocytotic recycling (1). Aβ monomers aggregate into Aβ fibrils, causing amyloid plaques in brain parenchyma and vasculature (4). Aβ activates microglia and astrocytes, inducing the complement system, local inflammatory responses and oxidative stress (5). CR 1 is the receptor of the complement C 3 b protein and participates in the clearance of Aβ from circulation (6). Besides causing increased Aβ endocytosis into glial cells, CLU is involved in Aβ clearance at the blood– brain barrier (7). APOE enhances amyloid plaque formation by conformational changes of Aβ. Clusterin (APOJ) and APOE are the main escorting proteins of Aβ in brain (7). Both are also important in cholesterol metabolism at the neuronal membrane (8) and high intracellular cholesterol may enhance APP amyloidogenic processing (2), which in turn can lead to membrane damage (9). Moreover, impaired cholesterol metabolism may influence synaptic dysfunction (10). Both PICALM and DNMBP are related at the synapse (10). Interaction of Aβ oligomers at the membrane is further connected to the calcium hypothesis in AD (11). Polymorphisms in the Ca 2+ channel CALHM 1 impair Ca 2+ permeability at the plasma membrane (11). In addition, PSENs function as ER Ca 2+-leak channels and several early-onset mutations impair Ca 2+-leak-channel function, resulting in an excessive Ca 2+ accumulation in the cytosol. An excessive Ca 2+ is taken up by mitochondria, further leading to oxidative stress and apoptosis (12). http: //hmg. oxfordjournals. org/content/19/R 1/R 4/F 2. expansion. html
Genetic Diagnosis and Counseling
Genetic testing (also called DNA-based tests) is among the newest and most sophisticated of techniques used to test for genetic disorders which involves direct examination of the DNA molecule itself. Other genetic tests include biochemical tests for such gene products as enzymes and other proteins and for microscopic examination of stained or fluorescent chromosomes. Genetic tests are used for several reasons, including: - carrier screening, which involves identifying unaffected individuals who carry one copy of a gene for a disease that requires two copies for the disease to be expressed - preimplantation genetic diagnosis - prenatal diagnostic testing - newborn screening - Genealogical DNA test (for genetic genealogy purposes) - presymptomatic testing for predicting adult-onset disorders such as Huntington's disease - presymptomatic testing for estimating the risk of developing adult-onset cancers and Alzheimer's disease - confirmational diagnosis of a symptomatic individual -forensic/identity testing The results of genetic tests are not always straightforward, which often makes them challenging to interpret and explain. May not always be correct. When interpreting test results, healthcare professionals consider a person’s medical history, family history, and the type of genetic test that was done.
Amniocentesis is a procedure for obtaining fetal cells for genetic testing. The diagram below shows the steps in the amniocentesis procedure for prenatal tests.
Chorionic villus sampling (CVS) is a form of prenatal diagnosis to determine chromosomal or genetic disorders in the fetus. It entails sampling of the chorionic villus (placental tissue) and testing it for chromosomal abnormalities, usually with FISH or PCR. CVS usually takes place at 10– 12 weeks' gestation, earlier than amniocentesis (14– 16 weeks). It is the preferred technique before 15 weeks. Risk of miscarriage in CVS in about 0. 5 - 1%. (the risk is 0. 6 – 0. 8% in amniocentesis; but it can be fatal if performed before 15 wks).
MULTIPLEX PCR FORENSIC OR PRENATAL GENETIC SCREENING PURPOSES DNA forensics in criminal investigations can use multiplex PCR with comparative human samples to assign identity in criminal cases as shown in the example below. These data would indicate high probability that the suspect sexually assaulted victim 2.
Preimplantation genetic diagnosis (PGD) is a method used to genotype human embryos for known genetic defects prior to uterine implantation. Removal of a single cell at the 8 cell stage is can be done in the laboratory to provide material for PCR analysis. Selected embryos are then re-implanted into the womb. In this hypothetical example, the Factor VIII gene defect is carried on one of the two X chromosomes on the maternal side. PEP/PCR amplification is used to randomly amplify the entire genome. Amplification of the ZFX and ZFY regions of the X and Y chromosomes, respectively, provides is diagnostic test for embryo gender.
IVF (in vitro fertilization) has led to this procedure, allowing parents to weed out genetically defective embryos. This procedure is called preimplantation genetic diagnosis (PGD). PGD is available for a large number of monogenic disorders, that is, a condition is due to a single gene only, (autosomal recessive, autosomal dominant or X-linked disorders) or a chromosomal structural aberration (such as a balanced translocation). PGD helps these couples identify embryos carrying a genetic disease or a chromosome abnormality, thus avoiding diseased offspring. The most frequently diagnosed autosomal recessive disorders are cystic fibrosis, Beta-thalassemia, sickle cell disease and spinal muscular atrophy type 1. The most common dominant diseases are myotonic dystrophy, Huntington's disease and Charcot-Marie-Tooth disease; and in the case of the X-linked diseases, most of the cycles are performed for fragile X syndrome, haemophilia A and Duchenne muscular dystrophy. Though it is quite infrequent, some centers report PGD for mitochondrial disorders or two indications simultaneously.
Pharmacogenetics refers to a molecular genetic approach to the personalized prevention, diagnosis and treatment of disease. In this scenario, genotype fingerprinting would be performed early in life to identify gene mutations known to be associated with an increased risk of lung cancer. Early detection methods for lung cancer would include routine PCR-based DNA microchip analysis of lung fluid (sputum) to detect rare cancer cells that would be indicative of disease, and blood samples would be analyzed to detect tumor metastasis. http: //www. biochem. arizona. edu/classes/bioc 471/pages/Lecture 24. html
The National Society of Genetic Counselors (NSGC) officially defines genetic counseling as the process of helping people understand adapt to the medical, psychological and familial implications of genetic contributions to disease. This process integrates: 1) Interpretation of family and medical histories to assess the chance of disease occurrence or recurrence. 2) Education about inheritance, testing, management, prevention, resources and research. 3) Counseling to promote informed choices and adaptation to the risk or condition. A genetic counselor is an expert with a Master of Science degree in genetic counseling. In the United States they are certified by the American Board of Genetic Counseling. In Canada, genetic counselors are certified by the Canadian Association of Genetic Counsellors. Most enter the field from a variety of disciplines, including biology, genetics, nursing, psychology, public health and social work. Genetic counselors should be expert educators, skilled in translating the complex language of genomic medicine into terms that are easy to understand.
Term assignment (will count for a number of questions in your 5 th committee exam - the exact number to be determined) Below are some proteins important for normal functioning of the body. Each student will choose one from the list, and prepare a project, describing the life of this protein (from gene to transcription, splicing, translation, modifications, folding, sorting, secretion etc, if applicable, and if it’s a growth factor or hormone, also explaining where the receptors for this protein are expressed, and what signal transduction events take place in the target protein when this protein binds to its target receptor, the developmental effects or changes in cell behavior, any genetic mutations known for this gene, mode of inheritance if known, and its consequences, etc). Max of 6 students per protein – Asst. Nese Aysit will collect names. The project due date will be before the committee exam, to be announced later. 1) 2) 3) 4) 5) Insulin Leptin Prion protein Somatostatin PINK 1 6) thrombin 7) DRD 4 (dopamine receptor) 8) BRCA 1 9) caspase 8 10) alpha-1 antitrypsin 11) VP 1 (foot and mouth disease) 12) RUNX 2 13) melatonin 14) XPF 1 15) sphingomyelinase PLEASE NOTE THAT PLAGIARISM (from the internet, from a book, from a friend etc) WILL NOT BE TOLERATED. YOUR PROJECT MUST BE YOUR OWN WORK, YOUR OWN WORDS !
FORMAT OF THE PROJECT ASSIGNMENT The project will be written in review format and will include: 1) 2) 3) 4) 5) A cover page (name of the student, title of the project / protein, date project is delivered) An introduction Subsections as necessary A conclusion REFERENCES !!!!!!! Any material used in the project should be appropriately cited, and indicated in text. Sentences should not be exactly copied to your project, but rephrased in your own words. If the sentence has to be used exactly as it appears in the reference, than use quotation marks. List the references in alphabetical order (of the first author name). Document should not be longer than 4000 words (excludin references). See the sample review paper.