Hiperkineziler Tremorlar Kore Atetoz Hemiballismus Distoni Miyoklonus Tikler


Hiperkineziler • • Tremorlar Kore Atetoz Hemiballismus Distoni Miyoklonus Tikler Stereotipiler

Kore • İstem dışı, rastgele ve vücudun farklı kısımlarında ani spazmodik, hızlı, kısa süreli, düzensiz kas sıçramalarıdır • Bazen uzuvlarda zorlu hareketler olabilir • Dilde ve yüzde görülebilir • El sıkma eylemi aralıklı kesintiye uğrar • Yürüme danseder gibi düzensiz, dengesizdir • Konuşma tonu ve temposu patlayıcı olabilir

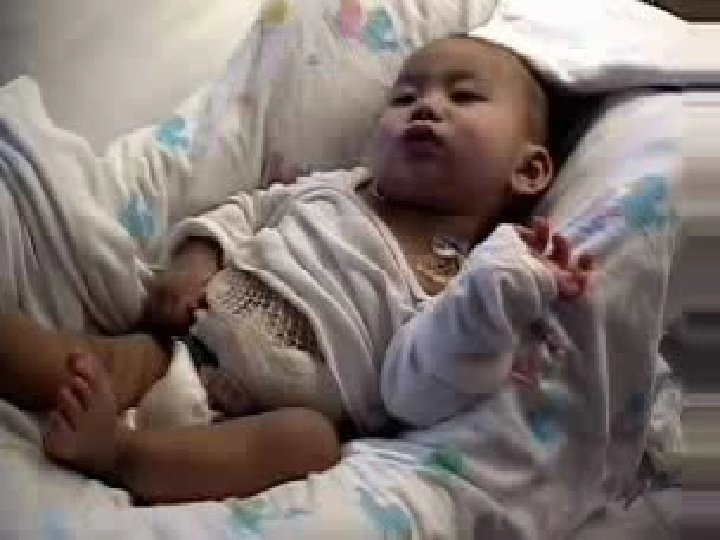
• İstemsiz, bir uzuvdan diğerine hızla yayılan, vücudun rastgele her yerinde, kısa süreli ve düzensiz kasılmalarıdır, tonus normal/ azalmıştır – Konuşma patlayıcı tarzdadır – Yutma ve solunum kasları etkilenir – Yürüme danseder gibi düzensizdir • İstemli hareket sürdürülemez, aralıklı kesintiye uğrar – Dil dışarıda tutulamaz, el sıkma, sabit bakış Jeneralize koreiform hareketleri olan Huntington hastası

Hızl ı n o l k o y k i i M T us u n o l k re o y o i K Rasgel M s e e r Ko i n o t s i D i n o t s k i i D T Yava ş Stereotip ik

Kore • Sydenham koresinde daha hızlı • Huntington hastalığı erken evresinde daha yavaş • Tardif diskinezide daha tekrarlayıcı ve stereotipiktir • Sıklıkla distoni / parkinsonizmle birlikte görülebilir – Distoniyle birlikte ise koreoatetoz adını alır Ballismus • Büyük amplitüdlü, hızlı, düzensiz, proksimal hareketlerin baskın olduğu koredir • Sıklıkla jeneralize, ancak fokal, segmental ya da bir beden yarısında olabilir

Bazal gangliyonlarda motor döngü • Direkt yol korteksten gelen uyarılar doğrultusunda motor programı aktive eder • İndirekt yol harekete odaklanır ve uygun hareketleri seçer • Bu kurallar döngünün her seviyesinde geçerlidir • Bir yol üzerinde herhangi bir seviyedeki aksama benzer hareket

Korenin Oluş Mekanizması Kaudat/subtalamik çekirdeklerin işlev bozukluğu İndirekt yolda striatum- dan GPe üzerine olan inhibitör etki azalır GPe serbestleşir, STN’u aşırı inhibe eder STN veri çıkış merkezle-rini yeterince uyaramaz ve talamus baskılanamaz Talamus istemsiz hareketleri gereği kadar inhibe edemez Kortek s İndire kt yol k Direk t yol T Hareket örneği jeneratör ü

Kore Nedenleri-I • Kalıtsal hastalıklar nörodejeneratif – Otozomal dominant • Fahr hastalığı • Huntington benzeri hast. 1, 2, 4 • Nöroferritinopati • Paroksismal KKA, PDKA • Selim kalıtsal • Spinoserebellar § Otozomal resessif § Beyin demir birikimi § Huntington benzeri has § Nöroakantositoz § Nöronal lipofüsinozis § Pantotenat kinaza ilişki n X-e bağlı resessif § Mc Leod sendromu n Mitokondriy el§ Leigh, . .

Fahr hastalığı • Striatal, pallidal, dentat, talamik, kapsüler ve subkortikal kalsifikasyon • 1/3 asemptomatik • Semptomatiklerin yarısında hareket bozukluğu • Herediter veya sporadik • Normallerin %1’inde de olan, MSS

Huntington Hastalığı Belirtileri • Kognitif Yıkım – Subkortikal demans – Epilepsi • Psikiyatrik belirtiler – İrritabilite ve agresyon – Apati, depresyon – Anksiyete, panik bozukluk – Psikoz aile ilişkileri bozulur • Hareket bozuklukları – Kore, miyoklonus, distoni – Serebellar ataksi – Parkinsonizm (Westphal) – Disfaji, boğulma tehlikesi, kilo kaybı, dizartri – Sakkadik göz küresi hareketlerini başlatma güçlüğü Aminoff, Greenberg, Simon. Clin Neurol, 1996
 4 p 16. 3 loküsünde (IT 15) i (1993) tekra rı")
Huntington Hastalığı (1872) 4 p 16. 3 loküsünde (IT 15) i (1993) tekra rı Huntingtin proteini toksik işlev kazanır

Huntington Hastalığı • OD geçişli, 4 p 16. 3 loküsünde CAG tekrarı • Prevalans 5/100 bin • Hastalık başlangıcı: 30 -50 yaşları, CAG tekrar sayısı arttıkça erken yaşta başlar • Serebral kortekste ve striatumda atrofi • GABA ve sentezini yapan GAD • Kore, psikoz, demans, epilepsi, serebellar ataksi, parkinsonizm (Westphal variantı) Aminoff, Greenberg, Simon. Clin Neurol, 1996

Huntington hastalığı tedavisi § Yavaşlatılamaz / durdurulamaz § Cerrahi tedavi: olgu bildirileri Coenzim Q 10, 600 mg/gün § GPi derin beyin yararlı stimülasyonu § Koreyi azaltan § Fötal kök hücrelerin bazal ilaçlar parkinsonizm gangliyonlara nakli Kore tedavisi üzerinde çalışılıyor (YE: subdural § Beyin dopamin düzeyini hem. !) § Tipik, atipik nöroleptikler Depresyon SSRI § Rezerpin, tetrabenazin Psikoz Nöroleptikler § Antikonvulsanlar Yutma § Levetirasetam, valproik asit, sorunu Jejunostomi karbamazepin Genetik bilgilendirme: İn § Glutamat antagonistleri § Amantadin vitro


Paroksismal Kinezijenik Diskinezi • Kore, atetoz, distoni ve ballismus atakları • Uzuvları, gövde ve yüzü tek/çift taraflı tutar, dizartri/mutizm olabilir • Bilinç: korunur • Harekete başlamak, ağır egzersiz, ürkme, hiperventilasyon ile tetiklenir • Sıklık: günde 100 – ayda 1 Süre: saniyeler- 5 dakika • Refrakter periyod: ~ 5 - 20 dakika • Duysal “aura”: karıncalanma hissi, kas gerginliği, baş dönmesi • Cinsiyet: 2 - 7 E / 1 K • Başlangıç yaşı: 6 -16 yaş ailevi / sporadik • Klinik seyir: Ataklar zamanla azalır veya tamamen kaybolur • Genetik heterojenite: 16 p 11 q 12; 16 q 13 -22. 1 infantil konvülsiyonların eşlik ettiği (ICCA) 16. kr.

16 yaş, erkek hasta, 6 yıldan beri günde 20 kez tekrarlayabilen, hareketle tetiklenen, istemsiz hareketleri mevcut

• • • Paroksismal Kinezijenik Olmayan Diskinezi DYT 8; 2 q 33 -35 Distoni, kore, atetoz ve ballismusu kapsar Uzuvlar, yüz, boyun ve gövde tek veya çift taraflı tutulabilir, bilinç korunur, ± dizartri Sıklık: günde 20 – yılda 1 Süre: 5 dakika-16 saat Duysal“aura”: kas gerginliği, parestezi, katılık Cinsiyet: E > K Başlama yaşı Erken başlangıçlı form: 2 ay< – Geç başlangıçlı form: 50 yaş< – Genellikle çocukluk veya büyüme çağı – • Klinik seyir: yaşla beraber kaybolabilir veya tedaviye yanıtsız olarak devam eder • Tetikleyen unsurlar: Uykusuzluk, alkol, kafein, yorgunluk, sıcak, soğuk, mens, heyecan, stres, çikolata

• Sıklıkla ilk 10 yılda başlar • Gelişim basamakları gecikebilir, dizartri olabilir • Kognisyon ve davranış korunur • İlerleyici değil • Ergenlik ve erken erişkin çağda kaybolabilir • Bazı durumlarda HH, miyoklonik distoni, primer distoni ve miyoklonuslu hastalar yanlışlıkla bu tanıyı alabilirler • Genetik olarak heterojen: OD bazı ailelerde kr. 14’teki TITF-1 geninde mutasyon

 • Japonya’da sık; Haw River Amerikan siyah")
Dentato-rubro-pallido-luysien atrofi (DRPLA - Haw River sendromu) • Japonya’da sık; Haw River Amerikan siyah ailesi • Kore, ataksi, miyoklonus, nöbetler, distoni, parkinsonizm, psikoz ve demans • Başlangıç 3. onyıl, sağkalım 20 yıl • 12. kr. ’da atrofin 1’i kodlayan gende kararsız artmış “CAG” trinükleotid tekrarı • Tipik intranükleer nöronal inklüzyonlar ubikütin ve

Nöroakantositoz • Orobukkal koreiform ve distonik hareket bozukluğu, nöbetler, demans ve davranış değişiklikleri, nadiren miyopati, aksonal nöropati, periferik yaymada dikensi akantositler • OR, 9 q 21 yerleşimli Ch. Ac geni mutasyonu(VPS 13 A) korein proteini • Başlangıç 4. onyılda, CPK kardiyomiyopati, hepatosplenomegali Mc. Leod sendromu • X-K geni mutasyonu, X’e bağlı resesif, 6. onyılda başlar


Kore Nedenleri-II • Otoimmün n Nörometabolik bozukluklar • Neoplazi n Sistemik metabolik – – – – – Sydenham, PANDAS n Amino asit metabolizma Enfeksiyon sonrası korebozuklukları SLE n Lipid metabolizma boz. (Niemann. Antifosfolipid antikor s. Pick, lizozomal depo hastalıkları) Gebelik koresi n Polisitemia vera Henoch-Schönlein n Porfiri, kernikterus, purin met. boz. Behçet hastalığı (Lesch-Nyhan sendromu) Poliarteritis nodoza n Tiamin / niasin eksikliği Multipl skleroz n Wilson hastalığı – Bazal gang. tutulumu – Paraneoplastik, lenfoma, metastaz • Vasküler: AVM, SVH • Çeşitli: Kalp cerrahisi, elektrik çarpm. , serebral felç Addison, Na, Ca, Mg Böbrek yetmezliği, üremi, Hepatik ansefalopati, hepatoserebral dejenerasyon n Hipertiroidi, hiperparatiroidi n Hipertiroidizm, hipoparatiroidi n Hipo / hiper -glisemi veya – natremi, n n n

Sydenham Koresi 1686 A grubu hem. streptokoklara karşı gelişen antikorlar beyin korteksi, bazal gangliyonlar ve serebellum dokusuyla çapraz reaksiyon yapar Nöronlarda kalmodulinin eşlik ettiği kinazların işlevi bozulur ARA tanısı Major: Kardit (valvülit ve mitral stenoza yol açabilir Her olguda ( eko) Poliartrit (gezici ), Kore, Eritema marginatum, Subkütan ( nodüller Minör: EKG (Uzamış R-R mesafesi), Artralji, akut faz ( reaktanları, Geçirilmiş RA’in kanıtı (Kızıl, Boğaz kültürü, ASO), ( Ateş Ta nı { 2 major veya 1 major 2 minör + enf. kanıtı

Sydenham Koresi § § § ARA’te %10 -30 sıklıkta; Kızlarda 2 kat daha sık 5 -15 yaşlarında görülür; jeneralize / hemikore İrritabilite ve OKB gibi kişilik değişiklikleri Romatizmal ateş-kore arası 2 -9 ay Kore 4 -6 hafta sürer %20 olguda tekrarlar, gebelik, doğum kontrol hapı ile geç tekrarlama § Sedimentasyon, CRP, bazen ASO § Antistreptokinaz, antinöronal, anti bazal gangliyon antikorlar § Romatik B hücresi allojeni D 8 / 17 (+) 2000 Gilroy. Basic Neurology,

Sydenham Koresi Tedavisi • Kendiliğinden düzelebilir, ancak kore özürlülük yaratacak şiddette ise tedavi edilir • Antikonvulsan ilaçlar: sodyum valproat • Dopamin antagonistleri: haloperidol, pimozid, . . . Potansiyel yan etki tardif diskinezi olabilir • Gerekirse kortikosteroidler, ya da IV Ig. G veya plazmaferez İkincil koruyucu tedavi • 21 günde bir benzatin penisilin G, i. m. (penisilin alerjisi varsa eritromisin) – Artrit veya kore geçirmişse 18 yaşına kadar – Romatizmal valvulopati gelişmişse ya da sağlıkçı, öğretmen gibi kalabalıkla temas içinde yaşıyor ve tekrar enfeksiyon -bilhassa endokardit- geliştirme olasılığı varsa ömür boyu

Antifosfolipid antikor sendromu SLE geç Syndenham 2. -5. aylar, bazen postpartum Kognitif değişiklik Semptomlar haftalar-aylar içinde kendiliğinden sonlanır


Kore Nedenleri-III n Sporadik selim § Enfeksiyöz n Fizyolojik süt çocukluğu § Creuzfeldt-Jacob hast. n Senil § AIDS § Difteri n Sporadik § Nörosifiliz nörodejeneratif § Lyme n Olivopontoserebellar § Ansefalit, lejiyoner n İlaçlara bağlı hast. , . . n Antiepileptikler § Toksinlere bağlı § Sarkoidoz, apse n Dopaminerjik ilaçlar § Etanol (zehirlen. /ani n Kokain, amfetamin, . . kesme) n Östrojen, lityum, § Karbon monoksit steroid, § Manganez digoksin § Civa n Trisiklik antidepresanlar § Talyum n Nöroleptikler § Toluen (yapıştırıcı)

Nöroleptiklere bağlı Tardiv Diskinezi


7 aylık bebek Herpes simpleks ansefaliti

Uzuv proksimalinde, yüksek Hemiballismu amplitüdlü, döndürücü/savurucu, s şiddetli hareketler q Uykuda kaybolur q. Subtalamik nükleus lezyonları: q İndirekt yol üzerinde globus pallidus interna/substansiya nigra retikulata yeterince uyarılamaz talamus gereği kadar inhibe olamaz ve serebral korteks aşırı uyarılır karşı beden yarısında hemiballismus q. Orta-ileri yaşlarda, sıklıkla q beyin-damar hastalıkları q ketozsuz hiperosmolar Jancovic&Tolosa, 1 hiperglisemi q

Sağ STN’ta hematomu olan 66 y, K Sol hemiballismus Sağ GPi pallidotomi

5 yıllık bir süre boyunca ardışık 51 kore olgusu: Piccolo ve ark. 2003 – % 40 vasküler (akut başlar, fokal bulgu verir) – % 20 Borelliozis, hiperglisemi, hipoksi, hiponatremi, nöroakantositoz, Sydenham, SLE – % 14 ilaçla tetiklenen – % 10 HIV ilişkili – % 10 Huntington hastalığı (aile öyküsü) – % 6 bilinmeyen

Laboratuvar İncelemeleri • • Tam kan sayımı polisitemia rubra vera Periferik kan yayması Nöroakantositoz Kreatin fosfokinaz Eritrosit sedimentasyon hızı vaskülit • Hiperkoagulasyon eğilimi – a. PTT, lupus antikoagulanı SLE – Antinükleer antikor – Antikardiolipin antikorlar Antifosfolipid sendromu, SLE } } • • Kan biyokimyası non-ketotik hiperozmolar hiperglisemi Tiroid fonksiyon testleri tirotoksikoz Serum kalsiyum, parathormon hipokalsemi, . . Bakır, seruloplazmin ve 24 st’lik idrarda bakır Wilson hastalığı ASO, CRP, anti-DNAaz B titreleri Sydenham koresi, 10%(-) Soy ağacı DNA analizi Huntington, SCA 1, 2, 17 ve DRPLA, . . . Gebelik testi, HIV, 24 st’lik idrar-BOS/organik amino asitler EEG, BT, MRG

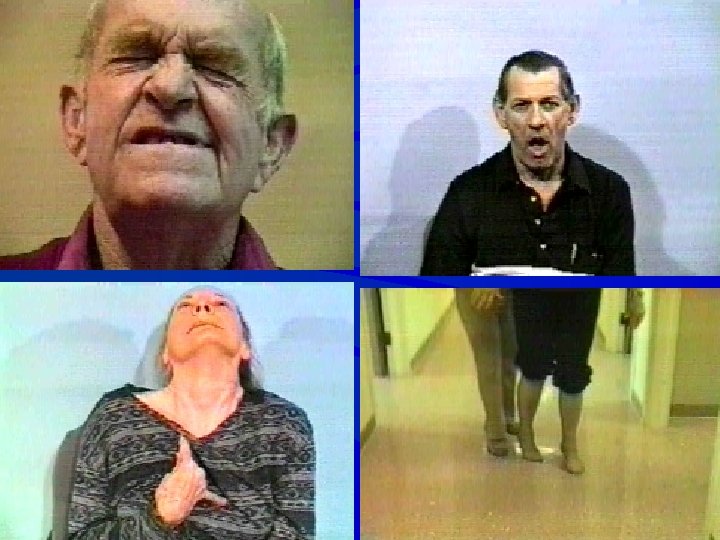
Distoni § Parkinson hastalığından sonra en sık görülen hareket bozukluğudur § İstemsiz gelişen sürekli kasılmaları etkiledikleri vücut bölgesini döndürür, tekrarlayıcı hareketlere / anormal duruş şekline yol açar § Agonist ve antagonist kaslar aynı anda kasılır

Distonik Kasılmalar § Yavaş veya hızlıdır ancak hareketi oluşturunca sürekli hal alır § Yönü sabittir ya da bir duruş biçimine yol açar § Bir ya da çok vücut bölgesini etkiler § İstemli hareket, stres & yorgunlukla artar § Dinlenme & uykuda azalır § Dokunma veya proprioseptif hilelerle azalır

Distoninin Fizyopatolojisi PET çalışmaları *DYT 1 taşıyıcılarında *DYT 1 hastalarında lentiform nükleus talamus serebellum suplemanter motor mezensefalonda kortekste artmış metabolik aktivite gösterir

Distoninin Fizyopatolojisi PET çalışmaları *DYT 1 taşıyıcılarında hastalarında talamus lentiform nük. serebellum suplemanter mezensefalonda motor kortekste artmış metabolik aktivite gösterir

• • • DİSTONİDE HAREKET TİPLERİ Miyoritmi Atetoz Torsiyon spazmı • Distonik tremor • Miyoklonik distoni

Distonilerin Sınıflandırılması • Başlangıç yaşına göre - Çocukluk: 0 -12 yaş - Gençlik : 13 -20 yaş - Erişkin : 20 < yaş • Etyolojiye göre - Primer - Distoni “artı” - Sekonder (semptomatik) - Heredodejeneratif hastalıklar Merrit’s Neurology, 2000

CTF HAREKET BOZUKLUKLARI BİRİMİ DİSTONİ SERİSİ Hülya Apaydın, Sibel Özekmekçi

Distonilerin Sınıflandırılması Vücut dağılımına göre - Fokal - Segmental *Kraniyal *Aksiyal *Brakiyal *Krural - Mültifokal - Jeneralize - Hemidistoni

Primer Distoniler Primer jeneralize 12. 5 8. 2 “Zirve” yaşları Başlangıç Seyir %95 bir kol/bacak Jeneralize (%50 -90) Primer fokal 45 Üst taraf Fokal/segmental Progresyon Hızlı Spontan remisyon Seyrek Sık OD OD Kalıtım şekli Penetrans Gen Mutasyonu %30 -40 Yavaş ~%25 DYT 1 DYT 2, DYT 4 (9 q 34, GAG delesyonu) DYT 6 (8 p 21 q 22) ( DYT 7(18 p) DYT 7(
 ve olmayan (NJ) bireylerde DYT 1 -% 82 Erken-başlangıçlı DYT")
Askenazi Yahudisi olan (AJ) ve olmayan (NJ) bireylerde DYT 1 -% 82 Erken-başlangıçlı DYT 1 - DYT 1 atalardan kalıtılmış 1 -9 10 -19 20 -29 30 -39 40 -49 50 -59 60> Yaşlar - % 53 Erken başlangıçlı DYT 1 vericiden çok çoğul DYT 1 mütasyonları - Yüksek oranda erken başlangıçlı olgular DYT 1 GAG eksilmesine bağlı değil - Erken başlangıçlı DYT 1 olmayanlar DYT 1 gibi davranıp yaygın distoniye dönüşebilir 1 -9 10 -19 20 -29 30 -39 40 -49 50 -59 60> Yaşlar

# ●: primer DYT 1 Loküs 9 q 34 DYT 2 DYT 3 Xq 13. 1 DYT 4 - Kalıtım Fenotip Gen ürünü OD Erken uzuv başlangıçlı PTD Torsin A OR OD Erken başlangıçlı Çingenelerd Filipin-distoni e Lubag parkinsonizm Fısıltılı (adduktor) disfoni XR - DYT 5 14 q 22. 1 OD DRD / parkinsonizm GCH 1 DYT 6 8 p OD Karışık - DYT 7 18 p OD Erişkin servikal - DYT 8 2 q 33 -35 OD PDC/PNKD - DYT 9 1 p 21 OD - DYT 10 16 OD Epizodik koreoatetoz / ataksi + spastisite PKK / PKD (EKD 1 & 2) DYT 11 7 q 21 OD Miyoklonus distoni DYT 12 19 q OD DYT 13 1 p 36 OD Hızlı başlangıçlı distoni parkinsonizm Servikal / kraniyal / brakiyal Sarkoglikan - -

Kranioservikal Distoniler § Spazmodik tortikollis § Anterokollis § Retrokollis § Laterokollis § Blefarospazm § Oromandibüler distoni § Meige sendromu § Larenjeal distoni

k k Birçok hastada bu postürler birarada görülür

Boyun Anatomisi: “Ön üçgen”

Boyun Anatomisi: “Arka Üçgen”

Boyun Anatomisi: Arka Grup Kaslar
, ansefalit,")
Sekonder Distoni İşaretleri Merrit’s Neurology, 2000 • Etiyolojik faktör öyküsü : travma (kafa/periferik), ansefalit, toksine maruz kalma, doğumda anoksi • Nörolojik anormallik bulgusu: demans, nöbetler, ataksi, parezi, spastisite, amiyotrofi, göz bulgusu, parkinsonizm • İstirahatte başlama • Konuşma bozukluğunun erken başlaması • Hemidistoni / Erişkinde bacak distonisi • Anormal beyin görüntülemesi/Lab. bulgusu • Yalancı güçsüzlük vs. gibi psikojen etyoloji

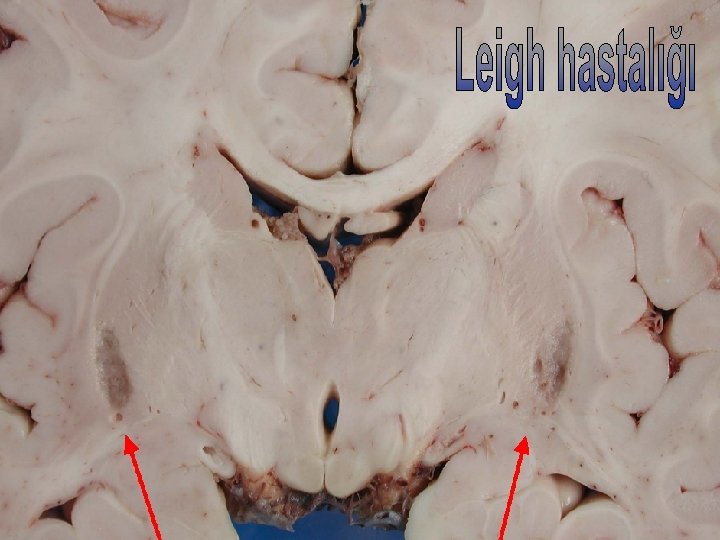
Distoniye Yol Açan Heredodejeneratif Hastalıklar Weiner WJ, Lang AE. Mov. Disord 1989 • Metabolik Bozukluklar • Dejeneratif Bozukluklar Wilson hastalığı Parkinson hastalığı Aminoasidüriler Mültipl sistem atrofi Metakromatik lökodis. Huntington hastalığı PSF Seroid lipofüsinozis Kortikobazal gang. dej. Jüvenil distonik lipidoz Lubag Gangliyosidozlar Pallidal dejenerasyon Lesch-Nyhan sendr. Hallervorden Spatz Leigh hastalığı Nöroakantositoz Mitokondriyel hastl. Ataksi telanjiektazi Ailevi bazal gang. kal. Hızlı baş. distoni-Pizm

Hallerworden-Spatz

Sekonder Distoni Nedenleri § Perinatal beyin hasarı: Atetoid beyin felci, geç başlay. distoni, pakigiyri § Ansefalit, enfeksiyonl. postenf: Reye sendr, SSPE, arı sokması, HIV, Creutzfeldt-Jacob, § Kafa travması § Talamotomi § Pr. antifosfolipid send. § Fokal beyin dam. hasarı § AVM § § § Hipoksi Beyin tümörü Arka çukur tümörleri Beyin sapı lezyonu (pontin miyelinoz) Servikal med. sp lezy. Lomber kanal stenozu Periferik hasar Elektrik çarpması Mültipl skleroz İlaç/toksinlere bağlı Metabolik/psikolojik

Nöroleptiklere bağlı Tardiv Diskinezi Distoni

Dopamin Reseptörlerini Bloke Ederek Sekonder Distoni Yapan İlaçlar § Asetofenazin § Piperasetazin § Amoksapin § Proklorperazin § Klorpromazin § Promazin § Flufenazin § Prometazin § Haloperidol § Tietiylperazin § Loxapin § Tioridazin § Mezoridazin § Tiotiksen § Metaklopramid § Trifluoperazin § Molindon § Triflupromazin § Perfenazin wemove. org, 2001

Çocukluk Çağı Akut Erişkin Dönemi Kronik Akut İlaçlar Keiser-Fleischer Simetrik Asimetrik i. v. Akineton İlaçlar Vasküler ile düzelir Wilson Var Yok Huntington Spazmodik Wilson Hast. Yok Diğer nörolojik bulgu Var MR (+) Yok İTD MR (-) Fokal distoni Dopa yanıtlı distoni Lokalizasyon Blefarospazm OMD disfoni Yazıcı krampı Spazmodik tortikollis Aile Öyküsü Var Kronik Perinatal Posttravmatik Ansefalitik İlaçlar Kafa travması Toksinler

DYT 5: Dopa-yanıtlı Distoniler Jancovic, Tolosa. PD&Mov Disord, 1998 • OD geçişli, 14 q, GTP-CH 1 geni • En sık 4 -8 yaşında (kız/erkek: 2 -3/1) • Distoni alt ekstremiteden başlar erişkin çağda parkinsonizm gelişir • Diurnal variyasyon: sabah semptomsuz • DA nöronları sağlam, sentezi bozuktur • L-dopa yarar sağlar/uyku yarar sağlar • OR, 11 p 15, Tirozin hidroksilaz (TH) geni • Ayak distonisi, 40 yaş >parkinsonizm • L-dopa ihtiyacı giderek diskinezi gelişir
, OR kofaktörü BH 4 LEVODOPA BH")
DOPA-YANITLI DİSTONİLER TİROZİN Tirozin hidroksilaz (11 p 15), OR kofaktörü BH 4 LEVODOPA BH 4 (tetrahidrobiyopterin) biyosentezinde 1. basamağı katalize eden GTPCH 1 “guanozin trifosfat siklohidrolaz 1” geninde heterozigot mutasyonlar (14 q) OD 2. basamağı katalize eden 6 -piruvoyltetrahidropterin sentaz eksikliği
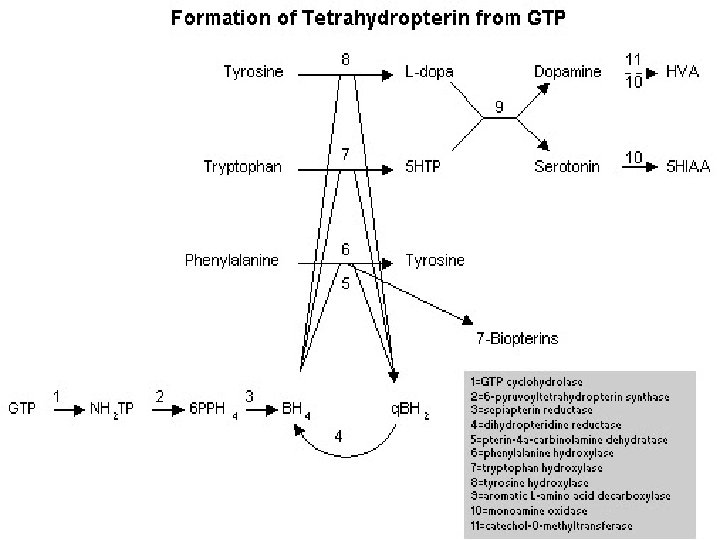


Fenilalanin Yükleme Testi • Tirozin hidroksilaz • Triptofan hidroksilaz & • Fenilalanin hidroksilaz’ın kofaktörü olan tetrahidrobiopterin kusurunu belirlemek üzere yapılır • DYD olan hastalarda anlamlı derecede yüksek fenilalanin/tirozin oranı vardır
 <5 (0 -25) 2 -20 OD, sporadik")
PKD PNKD PED 5 -15 (1 -35) <5 (0 -25) 2 -20 OD, sporadik OD Başlama yaşı Kalıtım Atak süresi <5 dakika Dakikasaatler 5 -30 dakika Atak sıklığı 100/gün; 1/ay 1/gün; 1/ay Eşlik eden özellikler Distoni, kore, epilepsi Asimetri ++ +++ 3/gün; 2/ yıl Kore, distoni, ataksi + +++ Atakları baskılayabilme Distoni, kore Tetikleyici, kolaylaştırıcı unsur Ani hareket, irkilme, hipervantilasyon, yorgunluk, stres Alkol, kafein, Uzamış egzersiz, heyecan stres, kafein, yorgunluk Tıbbi tedavi DFH, KMZ, FB, Klonazepam,

DİSTONİ TEDAVİSİ GENEL YAKLAŞIMLAR • Primer DT : semptomatik • Sekonder DT: altta yatan nedene yönelik • Medikal tedavide deneme-yanılma yöntemi uygulanır • Tedavi seçiminde yaş, anatomik dağılım, DT şiddeti belirleyicidir • Cerrahi tedavi son seçenektir

DİSTONİ TEDAVİSİ Dopaminerjik ilaçlar: DYD’de etkili, Jeneralize DT’li çocuklarda ilk ilaç Erişkin DT’lerinde denenebilir • Antidopaminerjik ilaçlar: Yan etki tardif diskinezi • Antikolinerjikler Jeneralize DT ilk 5 yıl/diğer DT biperiden 40 mg/gün, triheksifenidil 80 -120 mg/gün i. v. akut yarar varsa oral ted’den yarar görecektir • Benzodiazepinler • Baklofen: 40 -180 mg/gün , seçilmiş olgularda i. t. • Lidokain/Meksiletin • Antikonvülsanlar • Botulinum A toksini

Antidopaminerjik ilaçlar • Atipik nöroleptikler: klozapin, ketiapin • Dopamin depletörleri: tetrabenazin, rezerpin • Nöroleptikler : haloperidol, pimozid vb.

Distoni Cerrahisi Ablasyon ameliyatları: • Talamotomi: VİM nükleusuna/DYT 1 mutasyonu varsa iyi sonuçlar • Pallidotomi: Gpi, PV kısmına, bilateral. Yararlı etki 3. ay başlar, zamanla artar Periferik cerrahi: BFS, tortikollis, spazmodik disfonide

- Slides: 79