HEREDITARYACQUIRED HEMOLYTIC ANEMIA HEMOLYTIC ANEMIAS Hemolytic anemias reduced


 A. Hereditary 1. Membrane")













 resulting cytoskeleton")























 Alloimmune haemolytic")








 • PLATLET COUNTS < 20, 000 • MORE COMMON IN WOMEN")



























- Slides: 79
HEREDITARY/ACQUIRED HEMOLYTIC ANEMIA
HEMOLYTIC ANEMIAS Hemolytic anemias = reduced red-cell life span
Classification of Hemolytic anemias I. Red cell abnormality (Intracorpuscular factors) A. Hereditary 1. Membrane defect (spherocytosis, elliptocytosis) 2. Metabolic defect (Glucoze-6 -Phosphate. Dehydrogenaze (G 6 PD) deficiency, Pyruvate kinase (PK) deficiency) 3. Hemoglobinopathies (unstable hemoglobins, thalassemias, sickle cell anemia ) B. Acquired 1. Membrane abnormality-paroxysmal nocturnal hemoglobinuria (PNH)
II. Extracorpuscular factors A. Immune hemolytic anemias 1. Autoimmune hemolytic anemia - caused by warm-reactive antibodies - caused by cold-reactive antibodies 2. Transfusion of incompatible blood B. Nonimmune hemolytic anemias 1. Chemicals 2. Bacterial infections, parasitic infections (malaria), venons 3. Hemolysis due to physical trauma - hemolytic - uremic syndrome (HUS) - thrombotic thrombocytopenic purpura (TTP) - prosthetic heart valves 4. Hypersplenism
SOME TYPES OF HHA eg. • • SICKLE CELL DISEASE THALASSEMIAS G 6 PD DEFICIENCY HEREDITARY SPHEROCYTOSIS
THALASSEMIAS • MICROCYTIC, HYPOCHROMIC, HEMOLYTIC ANEMIA • MOST COMMON IN AFRICAN, MEDITERRANEAN, MIDDLE EASTERN, & SOUTHEAST ASIAN DESCENT • MULTIPLE VARIANTS
THALASSEMIAS • CHARACTERIZED BY DEFECTIVE SYNTHESIS OF GLOBIN CHAINS, UNABLE TO PRODUCE NORMAL ADULT HEMOGLOBIN • TRAIT THOUGHT TO BE PROTECTIVE AGAINST MALARIA AS WELL
HEMOGLOBIN • NORMAL ADULT RBC CONSISTS OF 3 FORMS OF Hb: - Hb. A - 2 α and 2 β globin chains - Hb. A 2 – 2 α and 2 δ globin chains - Hb. F - 2 α and 2 γ globin chains • THALASSEMIAS α and β
THALASSEMIAS • TYPES OF DZ CHARACTERIZED BY DEFFERING EXTREMES OF ANEMIA • DEPENDS ON AMOUNT OF INEFFECTIVE ERYTHROPOIESIS AND PREMATURE DESTRUCTION OF CIRCULATING RBC’S • HYPOXIA IN SEVERE CASES
G 6 PD DEFICIENCY • MOST COMMON HUMAN ENZYME DEFECT • X-LINKED DISORDER • AFFECTS 15% OF U. S. BLACK MALES • DECREASE IN GLUTATHIONE LEVELS
G 6 PD DEFICIENCY • HEINZ BODIES SEEN ON PERIPHERAL BLOOD SMEAR • NEONATAL JAUNDICE 1 -4 DAYS AFTER BIRTH IN SEVERE VARIANTS • INCREASE INCIDENCE OF PIDMENTED GALLSTONES AND SPLENOMEGALY
G 6 PD DEFICIENCY • ACUTE HEMOLYTIC CRISIS DUE TO: - BACTERIAL/VIRAL INFECTION - OXIDANT DRUGS (SULFAMETHOXAZOLE) - METABOLIC ACIDOSIS (DKA) - RENAL FAILURE - INGESTION OF FAVA BEANS
G 6 PD DEFICIENCY • DIAGNOSIS – QUANTITATIVE ASSAY DETECTING LOW ENZYME • TREATMENT – SUPPORTIVE AND PREVENTATIVE
HEREDITARY SPHEROCYTOSIS • RBS MEMBRANE DEFECT • MOST COMMON HEREDITARY ANEMIA FROM PTS OF NORTHERN EUROPEAN DESCENT • AUTOSOMAL DOMINANT • MUTATIONS IN SPECTRIN AND ANKYRIN (MEMBRANE PROTEINS)
HEREDITARY SPHEROCYTOSIS • SPHEROCYTES – IN PERIPHERAL BLOOD SMEAR • SPHEROCYTES UNABLE TO PASS THROUGH THE SPLEEN • SEVERE CASES REQUIRE A SPLENECTOMY
HEREDITARY SPHEROCYTOSIS • NEONATAL JAUNDICE IN 1 ST WEEK OCCURS IN 30 -50% OF HS PTS • ANEMIA, SPLENOMEGALY, JAUNDICE, AND TRANSFUSIONS NEEDED VARY DEPENDING ON SEVERITY OF DZ
Hereditary microspherocytosis 1. Pathophysiology - red cell membrane protein defects (spectrin deficiency) resulting cytoskeleton instability 2. Familly history 3. Clinical features - splenomegaly 4. Laboratory features - hemolytic anemia - blood smear-microspherocytes - abnormal osmotic fragility test - positive autohemolysis test - prevention of increased autohemolysis by including glucose in incubation medium 5. Treatment - splenectomy
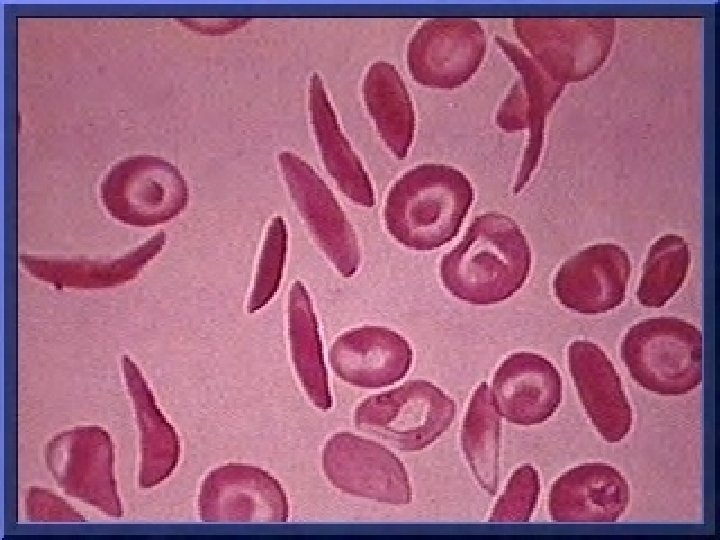
Sickle-Cell Anemia
Hemoglobin Composed of: 1 Heme and 4 Globin Chains 4 Types of Globin Chains: Alpha, Beta, Delta, Gamma
Sickle Cell Disease • Cannot make Beta Chains • Valine substituted for glutamate in 6 th position of beta chain
Sickle Cell Disease • Affects people of African descent • Affects 72, 000 people in the US • 2 million people are carriers • Occurs once in every 375 African American births
Sickle Cell Anemia Sickle Cell anemia is an inherited red blood cell disorder. Normal red blood cells are round like doughnuts, and they move through small blood tubes in the body to deliver oxygen. Sickle red blood cells become hard, sticky and shaped like sickles used to cut wheat. When these hard and pointed red cells go through the small blood tube, they clog the flow and break apart. This can cause pain, damage and a low blood count, or anemia.
The origin of the disease is a small change in the protein hemoglobin The change in cell structure arises from a change in the structure of hemoglobin. A single change in an amino acid causes hemoglobin to aggregate.
Hemoglobin is a carrier protein Hb. O 2 CO 2 Lungs deoxy Hb (CO 2) Tissues
Hemoglobin changes structure for efficient oxygen uptake and delivery Hb. O 2 deoxy Hb (CO 2) Strong binding state R state Weak binding state T state
The small change in hemoglobin structure leads to aggregation b Subunits Normal hemoglobin (Hb A) Sickle cell hemoglobin (Hb S)
Genetic Inheritance
Symptoms in Children • Start to appear at 6 months • Dactylitis (swelling of hands and feet) • Heart Enlargement • Growth Retardation • Delayed Sexual Development
Dactylitis
Sickle Cell Crisis • Severe pain caused by blocked blood flow • Triggered by Infection, Dehydration, Fatigue, or Emotional Stress • Can last up to 5 days
Splenic Sequestration • Spleen tried to remove abnormal cells • Becomes enlarged and causes pain • Autosplenectomy occurs • Usually not seen in adults
Symptoms of Anemia • Tiredness • Headaches • Dizziness • Faintness • Shortness of breath
Other Symptoms • Chronic, low-level pain in joints and bones • Abdominal pain • Retina Damage • Gallstones • Leg Ulcers (adults) • Chest pain
Leg Ulcers
Blood Picture • Sickle, Target and/or n. RBCs • Decreased Hemoglobin • Increased retic count • White cell count increased • WBC shift to the left
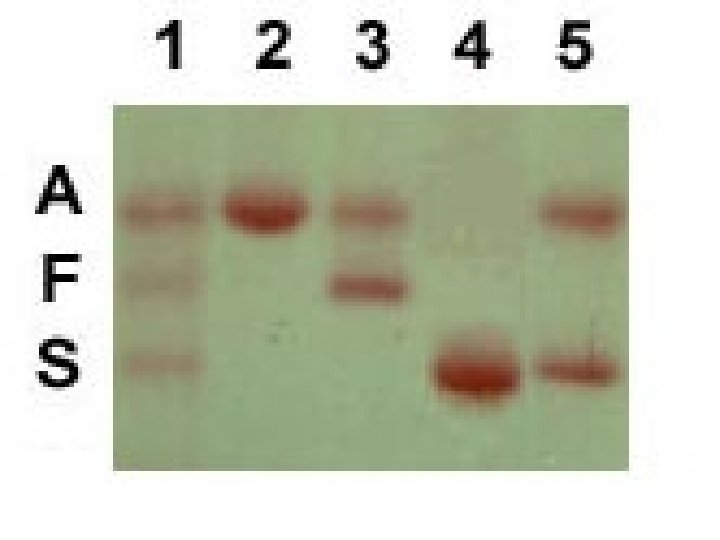
Hgb Electrophoresis • Amino acids in globin chains have different charges • Separates hemoglobin according to charge • 90% Hgb S, 10% Hgb F, small fraction of Hgb A 2
Prognosis • No cure • Life expectancy: 42 years men 48 years women • 85% reach the age of 20 • 50% reach age 50 • Causes of death: Infection, heart failure
Treatment • Pain Medication • Increase fluids • Blood Transfusion • Bone Marrow Transplant
Acquired Hemolytic Anemia
Introduction • • Increased RBC Destruction – Short RBC life span <120 days. Normocytic normochromic, reticulocytosis Anemia, Jaundice, marrow hyperplasia Splenomegaly, bilirubin gall stones Unconjugated “acholuric” (pale urine) Common types - AIHA, MAHA
Types of acquired HA • • Auto. Immune Haemolytic Anemias (+ve DAT) Alloimmune haemolytic anemias Drug-induced immune haemolytic anemias Red cell fragmentation syndromes Infections Chemical & physical agents Secondary Haemolytic anemias Paroxysmal Nocturnal Haemoglobinuria (PNH)
Pathogenesis of Jaundice: • • Hb Globin-Iron-Haem Bilirubin Glucoronide–Conjugation Bile Gut Stercobilinogen & Stercobilin (ex. in stool) Urobilinogen & Urobilin (ex. In urine)
Ketabolism of Hb:
Classification : • Auto Immune AIHA – Warm antibody type – Cold antibody type • Alloimmune – Transfusion reactions – Hemolytic disease of new born • Non-Immune – Microangiopathic hemolytic anemia – Infections – Malaria, clostridia, – Burns, Toxins, snake & spider bites.
Laboratory Diagnosis: • RBC Breakdown: – Hyperbilirubinemia – Urobilinogen, stercobilinogen – serum haptoglobins • RBC Production: – Reticulocytosis, *MCV – Marrow hyperplasia* • Damaged RBC – Morphology, fragility, survival
Laboratory Diagnosis: • Additional features of Intravascular Hemolysis: – Hb-naemia and Hb-nuria – Haemosiderinuria – Methaemoglobinemia
DRUG RELATED HA • ALPHAMETHYLDOPA • LEVODOPA • PROCAINAMIDE • SULFA DRUGS • • PENICILLIN CEFTRIAXONE CEFOTETAN QUINIDINE
MICROANGIOPATHIC SYNDROMES • THROMBOCYTOPENIC PURPURA • HEMOLYTIC UREMIC SYNDROME
TTP & HUS - PATHOPHYS • PLATELET AGGREGATION IN THE MICROVASCULATURE CIRCULATION VIA MEDIATION OF von WILLEBRAND’S FACTOR LEADS TO THROMBOCYTOPENIA AND FRAGMENTATION OF RBC’S AS THEY PASS THROUGH THESE OCCLUDED ARTERIOLES AND CAPILLARIES
THROMBOCYTOPENIC PURPURA (TTP) • PLATLET COUNTS < 20, 000 • MORE COMMON IN WOMEN AGES 1060 • FEVER, NEUROLOGIC DEFICITS, HEMORRAGE, AND RENAL INSUFFICIENCY • UNTREATED – 80 -90% MORTALITY
TTP • SCHISTOCYTES OR HELMET CELLS SEEN OF PERIPHERAL SMEAR • INCREASED BUN/Cr LEVELS
TTP • PREGNANCY IS THE MOST COMMON PRECIPITATING EVENT FOR TTP • PREECLAMPSIA SIMILAR TO TTP; DELIVERY TX FOR PREECLAMPSIA, NOT CURE TTP
TTP – ER TREATMENT • PREDNISONE 1 -2 mg/kg/day INITIALLY • PLASMA EXCHANGE TRANSFUSION IS FOUNDATION FOR TX (INFUSE FRESH FROZEN PLASMA IF TRANSFUSION UNAVAILABLE • AVOID PLATELET TRANSFUSION • NEVER USE ASPIRIN
TTP – ER TREATMENT • PT MAY NEED SPLENECTOMY • AZATHIOPRINE AND CYCLOPHOSPHAMIDE FOR THOSE WHO FAIL OR CANNOT TOLERATE STEROIDS
HUS • DZ OF EARLY CHILDHOOD • PEAK INCIDENCE BETWEEN 6 mo-4 yr • OFTEN FOLLOWS BACTERIAL/VIRAL ILLNESS • MORTALILY 5 -15%, WORSE IN OLDER CHILDREN & ADULTS
HUS • CHARACTERIZED BY -ACUTE RENAL FAILURE -MICROANGIOPATHIC HA -FEVER -THROMBOCYTOPENIA (NOT AS SEVERE AS TTP)
HUS • THE MOST COMMON CAUSE OF ACUTE RENAL FAILURE IN CHILDHOOD • E. Coli O 157: H 7 COMMON CAUSE • MICROTHORMBI ARE CONFINED MAINLY TO KIDENYS, WHERE TTP MORE WIDESPREAD
HUS – ER TREATMENT • MILD HUS < 24 hr OF URINARY SX NEELS ONLY FLUID/ELECTROLYTE CORRECTION AND SUPPORT CARE • STEROID THERAPY • HEMODIAYLSIS IF ACUTE RENAL FAILURE PRESENT • ABX TX CONTROVERSIAL WHEN E. Coli PRESNENT; DO NOT USE ANTIMOLITY DRUG, INCREASE RISK OF DEVELOP HUS
HELLP SYNDROME • HEMOLYSIS • ELEVATED LIVER ENZYMES • LOW PLATLET COUNTS
HELLP SYNDROME • 1 IN 1 OOO PREGNANCIES • SEEN IN PRESENCE OF ECLAMPSIA, PREECLAMPSIA, AND PLACENTAL ABRUPTION • MAY EXTEND UP TO 6 DAYS POSTPARTUM
HELLP SYNDROME • RUQ AND EPIGASTRIC PAIN – SEEN IN 90% OF PTS (POSSIBLE HEPATIC RUPTURE) • DX BASED ON LAB DATA • DECREASED SERUM HAPTOGLOBIN LEVEL MOST SENSITIVE
HELLP SYNDROME - TX • PROMPT DELIVERY OF INFANT • SUPPORTIVE CARE FOR SEIZURES AND HTN CRISIS • STEROIDS MAY HELP FETAL LUNGS, BUT NO BENEFIT TO HELLP SYNDROME
Warm AIHA • Idiopathic • Secondary Cold • Idiopathic • Secondary – Autoimmune lymphoma, drugs • • Spherocytes Ig. G antibody, C 3 d Direct Coombs - 37° Anti c / anti e – Infections – Lymphoma • • • RBC clumps Ig. M antibody Cold Ag. Titre 4° DAT +ve compl* Anti I / i
Warm • Idiopathic • Secondary – SLE, Autoimmune disorders. – CLL – Lymphoma – Drugs – Mdopa. AIHA Cold • Idiopathic • Secondary – Infections – Lymphoma • PCH (anti-P)
Warm AIHA • • Ig. G, C 3 d, rarely other Ab. Destruction in Spleen & RES Loss of partial membrane – spherocytes Clinical Features: – Spleenomegaly
Autoimmune hemolytic anemia caused by warm-reactive antibodies: I. Primary II. Secondary 1. acute - viral infections - drugs ( -Methyldopa, Penicillin, Quinine, Quinidine) 2. chronic - rheumatoid arthritis, systemic lupus erythemat. - lymphoproliferative disorders (chronic lymphocytic leukemia, lymphomas, WaldenstrÖm’s macroglobulinemia) - miscellaneous (thyroid disease, malignancy )
Autoimmune hemolytic anemia caused by cold-reactive antibodies: I. Primary cold agglutinin disease II. Secondary hemolysis: - mycoplasma infections - viral infections - lymphoproliferative disorders III. Paroxysmal cold hemoglobinuria
Alloimmune Haemolysis: • Antibody from another person. • Transfusion reactions • Haemolytic disease of Newborn (HDN) – RH-D, RH neg mother, + father, 2 nd baby Kleihauer test Hb. F, – ABO – Ig. G in O mother, mild*, 1 st babyagglutination & spherocytes, DAT neg/mild + • Post transplantation induced.
Paroxysmal nocturnal hemoglobinuria 1. Pathogenesis - an acquired clonal disease, arising from a somatic mutation in a single abnormal stem cell - glycosyl-phosphatidyl- inositol (GPI) anchor abnormality - deficiency of the GPI anchored membrane proteins (decay-accelerating factor =CD 55 and a membrane inhibitor of reactive lysis =CD 59) - red cells are more sensitive to the lytic effect of complement - intravascular hemolysis 2. Symptoms - passage of dark brown urine in the morning
3. PNH –laboratory features: - pancytopenia - chronic urinary iron loss - serum iron concentration decreased - hemoglobinuria - hemosiderinuria - positive Ham’s test (acid hemolysis test) - positive sugar-water test - specific immunophenotype of erytrocytes (CD 59, CD 55) 4. Treatment: - washed RBC transfusion - iron therapy - allogenic bone marrow transplantation
Microangiopathy:
Spherocytes:
AIHA Cold ab type: Clumping.
Assesment of HA Clinical features: - pallor - jaundice - splenomegaly
Laboratory features: 1. Laboratory features - normocytic/macrocytic, hyperchromic anemia - reticulocytosis - increased serum iron - antiglobulin Coombs’ test is positive 2. Blood smear - anisopoikilocytosis, spherocytes - erythroblasts - schistocytes 3. Bone marrow smear - erythroid hyperplasia
Diagnosis of hemolytic syndrome: 1. Anemia 2. Reticulocytosis 3. Indirect hyperbilirubinemia
Autoimmune hemolytic anemia diagnosis - positive Coombs’ test Treatment: - steroids - splenectomy - immunosupressive agents - transfusion