Hereditary diseases Congenital malformations Dr Gyrgy Fekete Congenital








 inheritance • Consanguinity increases the risk • The risk of two")









 • Elements of face are forming from the 4. embryonal")
 • Craniosynostosis (+ corpus callosum agenesia, hydrocephalus )")

 • Gene: 10 q")
")

 • Anatomical/ morphological deviation? •")
 Cytogenetics DNA analysis")
")
 • Edwards- syndrome (trisomy")

: lymphedema on the back of the hand / feet, pterygium")





")




 Prevalence: 1/ 2500 – 1/3000 Diagnostic criteria: 2 or")
, or at least one")
")









 • Genetic code of")
")
 • NO Lisch nodules,")
 syndrome, symptoms • • • Microcephalia, brachycephalia Deep anterior and")








 • Mild cognitive disturbances • Good")





, 1: 2500, CF transmembrane conductance")
 deficiency), 1: 10000, PAH gene")



 Semmelweis Egyetem, II. Sz.")


 Semmelweis Egyetem, II. Sz.")


 Recurrent")


 Small doses: no malformation High doses, continuous")
- Slides: 89
Hereditary diseases. Congenital malformations. Dr. György Fekete
Congenital malformations • • • Conception – organogenesis - birth Genetic causes Environmental factors (teratogens) Visible/ recognazible at birth Later manifestation
• Genetic conditions: causes of acute and chronic diseases • Onset of disease: fetus, infant, child, adult • Genetic abnormalities may produce: congenital malformations, metabolic disturbances, specific organ dysfunction, abnormalities of sexual differentiation
• Monogenic diseases: 1% of newborn babies • Chromosomal aberrations: 0. 5% • Multifactorial disorders: 1 -3%
• Monogenic diseases: 1% of newborn babies • Chromosomal aberrations: 0. 5% • Multifactorial disorders: 1 -3%
Inherited conditions • If a single allele has a detectable effect: dominant • If two functionally identical alleles cause the effect: recessive • XY males are hemizygous for genes on the X chromosome
Mendelian inheritance Autosomal dominant inheritance • If one parent displays a dominant condition and is heterozygous for the gene, each child has a 50% chance of receiving the single allele and of manifesting the condition • Not all individuals with the affected gene my be symptomatic • Penetrance: percentage of patients with the gene mutation who manifest symptoms
• Expressivity: spectrum of severity in patiens having clinical manifestation • Examples: achondroplasia, Crouzon syndrome, neurofibromatosis type I, II, Marfan syndrome, hereditary angioneurotic edema (HANE)
Autosomal recessive (AR) inheritance • Consanguinity increases the risk • The risk of two carriers of gene mutation having a child with AR diseases is 1 in 4 : 25% • Examples: phenylketonuria, cystic fibrosis, congenital adrenal hyperplasia, sickle cell disease, Gaucher disease, Pompe disease
Sex – linked inheritance • The gene locus is on the X chromosome • When the mother is a carrier of the gene mutation, 50% of male offspring will have the disease, and 50% of female offspring will be carriers • All daughters of the ill father will be obligate carriers • Examples: Duchenne muscular dystrophy (DMD), hemophilia A and B, adrenoleukodystrophy, Fabry disease
Mitochondrial diseases Semmelweis Egyetem, II. Sz.
Importance • Prevalence in newborns: 46 -50 %o • 25 -40 per cent of infant mortality • One factor in prematurity and intrauterine dystrophy • Severe conditions • Burden: child, family, society
Classification • Severity: major and minor malformations • Major: hindering significant organ functions – Isolated (GI atresias, Fallot – tetralogy) – Multiple: two or more organs, organ systems – Genetic: chromosome aberration, monolocus, other mechanism (uniparental disomy, genomic imprinting, triplet expansion, mitochondrial) – Teratogens (TORCH, chemicals, drugs, irradiation) – Genetic + environmental (multifactorial, complex diseases)
Minor malformations • Informative morphogenetic variants • Non - functional, harmless, esthetical deviations - Epicanthus
Supernumerary nipple
Minor malformations • Bifid uvula • 4 digits crease
Hypertelorism
Low – set ears
Craniofacial dysmorphy („peculiar face”) • Elements of face are forming from the 4. embryonal week. Face of fetus: 8. gestational week
Ossification anomalies of sutures (craniostenosis) • Craniosynostosis (+ corpus callosum agenesia, hydrocephalus )
Crouzon syndrome • AD, gene: 10 q 26
• Crouzon syndrome • Apert syndrome (Acrocephalosyndactyly type I) • Gene: 10 q 26, fibroblast growth factor receptor-2 (FGFR-2) • Advanced paternal age ( > 45 yrs) • Pfeiffer syndrome (Acrocephalosyndactyly type V) • Gene: 10 q 26, 8 p 11. 2 -11. 1(FGFR-1)
Splits • Split lip / palate (cheilo- gnatho- palatoschisis)
Mandibulofacial dysostosis: Treacher - Collins syndrome
Steps of examination • Parents, sibs, grandparents: resemblance? (photos!) • Anatomical/ morphological deviation? • Isolated or multiple? • Psychomotor retardation? • Other minor malformations? Hidden malformations (internal organs) ? • Teratogenic exposition? • Special methods / investigations • Councelling: prognosis, therapy
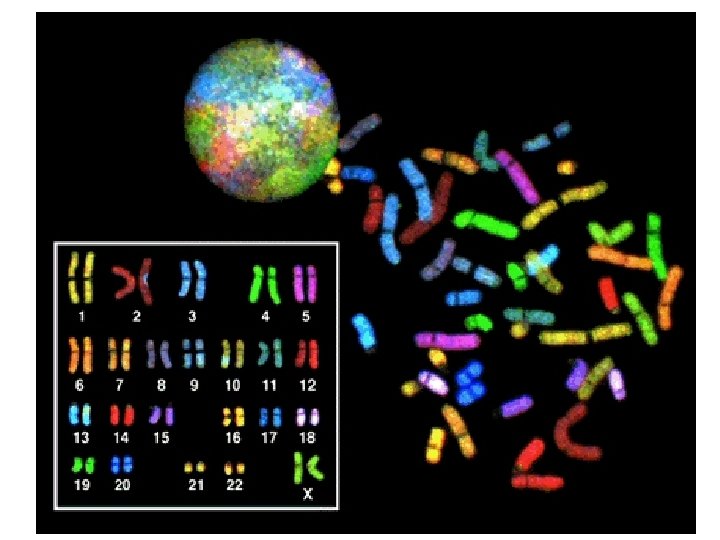
Special methods • • • Laboratory data Imaging techniques (CT, MRI) Cytogenetics DNA analysis Biochemical studies
Recognizable malformations in newborn age • Down- syndrome • ( trisomy chromosome 21 ) 21 q 22
Patau -, Edwards- syndrome • Patau- syndrome (trisomy chromosome 13) • Edwards- syndrome (trisomy chromosome 18)
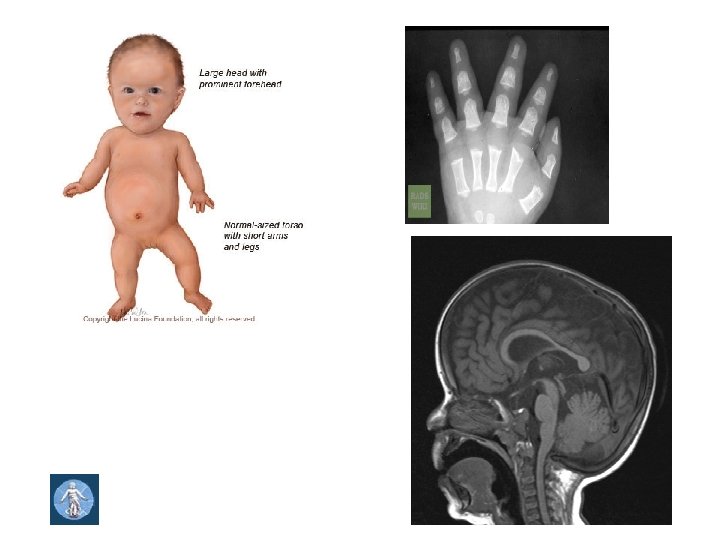
Prader – Willi syndrome
Turner syndrome (45, X): lymphedema on the back of the hand / feet, pterygium colli
Di. George syndrome
Isolated malformations / newborns • • • Anencephaly Spina bifida Hip dyslocation Atresias of esophagus and bowels Pyelectasy (obstructive uropathy) Diaphragma hernia Omphalokele Hirschsprung disease (megacolon congenitum) Congenital heart disease
Spina bifida
Congenital heart disease, pyloric stenosis
Club- foot, congenital dyslocation of the hip
Inguinal hernia , megacolon congenitum (Hirschsprung disease)
Retentio testis, hypospadiasis
Clinical signs and data of a possible chromosome aberration • Malformations, dysmorphisms of the skull and face (craniofacial dysmorphy) • Mental retardation • Multiorgan involvement • Maternal age: 35 yrs or more
Achondroplasia • 1: 5000, gene mutacions of fibroblast growth factor receptor-3 gene, 4 p 16. 3
Osteogenesis imperfecta
Neurofibromatosis type 1 (NF 1) Prevalence: 1/ 2500 – 1/3000 Diagnostic criteria: 2 or more of the following points are present 6 or more café- au -lait spots, diameter > 5 mm. (prepuberty), and > 15 mm. (postpuberty)
2 or more neurofibromas (fibromatous ( tumors of the skin), or at least one plexiform neurofibroma
Plexiform neurofibromas • potential for transformation into malignant peripheral nerve sheath tumors (malignant schwannomas)
Axillary and/ or inguinal freckling
Optic pathway glioma, spinal neurofibromas 2 or more melanocytic iris hamartomas (Lisch nodules )
Optic glioma precocious puberty , visual loss
Lisch nodules
Specific bone lesions Dysplasia of long bones , pseudoarthrosis of tibia, thinning of long t bone cortex
Vascular manifestations • Renal artery stenosis • Hypertension
First –degree family relative has a proven diagnosis of neurofibromatosis National Institutes of Health Consensus Conference 1987
NF 1 - symptoms of 59 male and 43 female pediatric patients Café-au-lait sp. Neurofibroma Plexiform neurofibroma Freckling Optic glioma Iris nodules Orthop. spine sympt. Other bone lesions Males % Females% 100 54, 2 100 34, 9 33, 9 59, 3 30, 5 39, 0 61, 0 28, 8 27, 9 65, 1 25, 6 32, 6 7, 0 Krumbholz A. et al. Monatsch Kinderheilk 2008, 156: 893 - 898.
Some infants and children present only with „café –au – lait” spots without any other neurofibromatosis symptom and sign (innocent spots): regular pediatric observation is needed macrocephaly (occipitofrontal circumference: >97. percentile) short stature (< 3. percentile), early dentition (< 5 mo. ) : suspected NF 1 new mutation complications: learning disabilities, mental retardation, tumors (CNS, neurofibrosarcoma)
NF 1 -gene , chromosome 17 (17 q 11. 2) • Genetic code of neurofibromin synthesis. Regulation of signal –transduction -protein RAS • GTPase activation • 61 exons • NF 1 gene mutations occur also in juvenile myelomonocytic leukemia, Watson syndrome, and breast cancer • 50% of cases are due to de novo mutations • Sporadic occurrence is associated with advanced paternal age
Differential diagnosis Healthy children: 19 / 1000 newborn babies (Michalk D, Schönau E, 1999) Ovarial cysts Juvenile xanthogranuloma
Legius syndrome • SPRED 1 mutations (15 q 13. 2) • NO Lisch nodules, neurofibromas, optic gliomas, bone lesions • Subcutaneous lipomas in adults • 7 coding exons
Cornelia de Lange (Brachmann) syndrome, symptoms • • • Microcephalia, brachycephalia Deep anterior and posterior hair border Synophrys (thick, meeting eyebrows) Ptosis, nystagmus, myopia Micrognathia long philtrum Thin lips, „carp” mouth Cheilo –gnatho - palatoschisis Malformations of limbs Hypoplastic penis, cryptorchism Mental retardation
Williams - Beuren syndromedeletion of elastin gene
K. M. female child Birth date: 02. 28. 2009. Presentation: 08. 23. 2010. Symptoms : • Somatic and psychomotoric developmental delay • Hypotonic muscles • Craniofacial dysmorphy
History • First child, healthy parents • Mother was 24, father 27 when she was born • Birth weight: 2640 g. , length: 45 cm, 39. gestational week, normal delivery , Apgar: 10/10
Craniofacial dysmorphy • • Sunken nasal bridge Epicanthal fold Long philtrum Prominent lower lip Hypodontia Mikrodontia Blue/ green iris
Hypertension • Supravalvular aortic stenosis • Peripheral pulmonary stenosis
Other symptoms • • Kyphoscoliosis, arthropathy Inguinal and umbilical hernia Loose skin Chronic constipation, diverticulosis Deep voice Sensoneural hearing loss Congenital malformations of kidneys • Laboratory: Hypercalcemia • Nephrocalcinosis
Endocrinological problems • • Hypothyreoidism Early puberty Early menarche Diabetes mellitus
Radioulnar synostosis
• Friendly, extroverted personality, („cocktail party personality”) • Mild cognitive disturbances • Good vocabulary • Good skill to music, singing
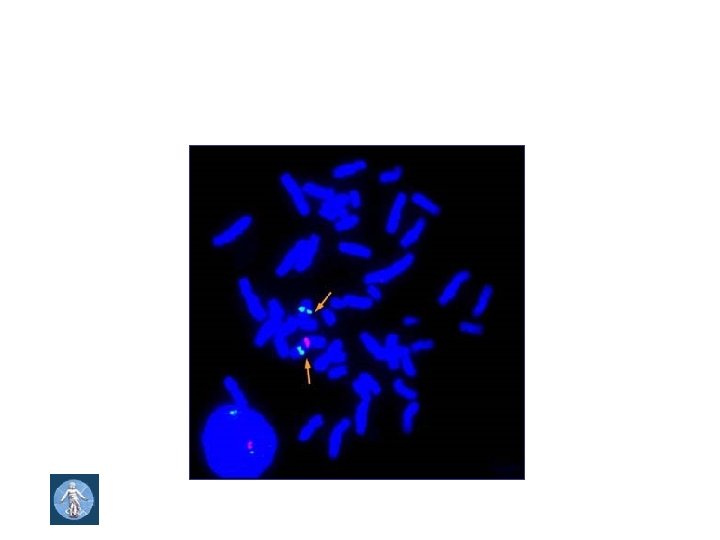
Williams- Beuren syndrome Incidence 1 : 8000 , sporadic, unbalaned meiotic cross - over • 7 q 11. 23 m. Mckrodeletion • Deletion of elastin gene and neighbouring genes • GTF 2 IRD 1 (General transcription factor II-I repeat domain- containing protein 1) • GTF 2 I (General transcription factor II-I) • Genetic diagnostic methods: - FISH - DNA analysis
• J. C. P. Williams, cardiologist, Auckland, New - Zealand, 1930 - ? Publication in: 1961 • Alois J. Beuren, cardiologist, Göttingen, 1919 -1984
Genetic counselling • • • Discussion of present and later symptoms Prognosis Pedigree analysis • Special care and support of skills • Patient organisations
Marfan syndrome • 1: 16000 -25000, fibrillin-1 gene mutations, 15 q 21. 1 Semmelweis Egyetem, II. Sz.
Syndrome • Defects of multiple tissues • Patients’ phenotypes are similar to each other • Characteristic presentation symptoms • Clinical diagnosis is feasible in most cases
Inborn errors of metabolism • Cystic fibrosis (CF, mucoviscidosis), 1: 2500, CF transmembrane conductance regulator (CFTR) gene mutations, 7 q 31. 2 (more than 2000 mutations) • Congenital adrenal hyperplasia (CAH, adrenogenital syndrome), 1: 5000 -8000, CYP 21 gene mutations , 6 p 21. 3 Semmelweis Egyetem, II. Sz.
• Phenylketonuria (PKU, classic type, phenylalanine hydroxylase (PAH) deficiency), 1: 10000, PAH gene mutations, 12 q 22 • Galactosemia, 1: 35000 -60000, GALT gene mutations, 9 p 13 • Biotinidase deficiency, 1: 24000, BTD gene mutations, 3 p 25 • Glycogen storage diseases (von Gierke) • Mucopolysaccharidoses (Hurler/Scheie) Semmelweis Egyetem, II. Sz.
Semmelweis Egyetem, II. Sz.
Cystic fibrosis Semmelweis Egyetem, II. Sz.
Cystic fibrosis Semmelweis Egyetem, II. Sz.
Robert Guthrie (1916 - 1995) Semmelweis Egyetem, II. Sz.
Tandem mass spectrometry Separation of molecules according to their charge/ mass relation following conversion of metabolites into ions Semmelweis Egyetem, II. Sz.
Semmelweis Egyetem, II. Sz.
Hurler disease (MPS I) Semmelweis Egyetem, II. Sz.
Pompe disease
Pompe disease
Clinical signs and data of monolocus hereditary diseases • • Multiorgan involvement (syndrome) Recurrent familial occurrence Consanguinity Characteristic phenotype
Reductional malformations of limbs • Amely
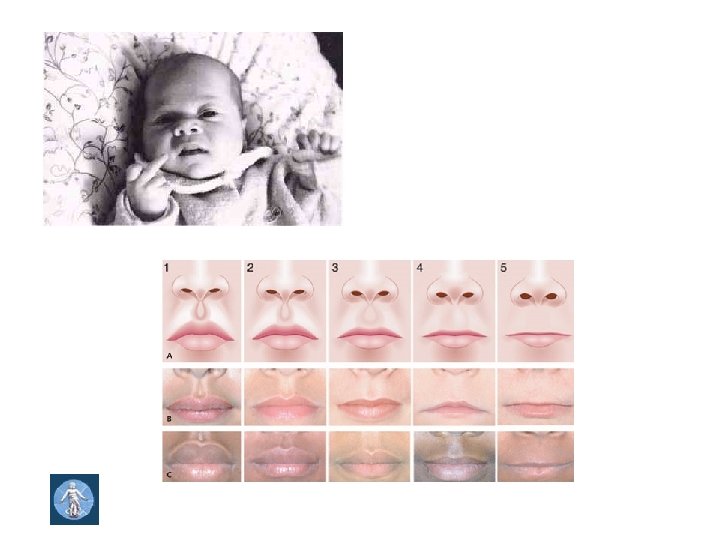
Fetal alcohol syndrome • • • Intrauterine and postnatal dystrophy, microcephaly Short, thin eye openings, epicanthus Deep nasal bridge Small nose Hirsutism on the frontal region Low-set ears, ear lobe deformities Flat os zygomaticum High palate, cheilo- gnatho- palatoschisis Thin upper lip, smooth philtrum Micrognathia
Drug abuse • Amphetamins (Ecstasy, metamfetamin/ice/, speed) Small doses: no malformation High doses, continuous use: cong. heart disease, cheilo- gnatho - palatoschisis Other teratogenic agents (alcohol, smoking, etc. )