Hereditary Clotting Factor Deficiencies Youngshil Park M D



")
 - milder than any of the three haemophilias")


 • • Extrinsic coagulation pathway screen Normal")



 = % desired (rise in FVIII) X body weight(kg) X")














 Factor V deficiency Congenital afibrinogenemia Factor")

- Slides: 33
Hereditary Clotting Factor Deficiencies Young-shil, Park, M. D. Division of Hematology-Oncology Department of Pediatrics East-West Neo medical Center Kyung Hee University
Hemophilia
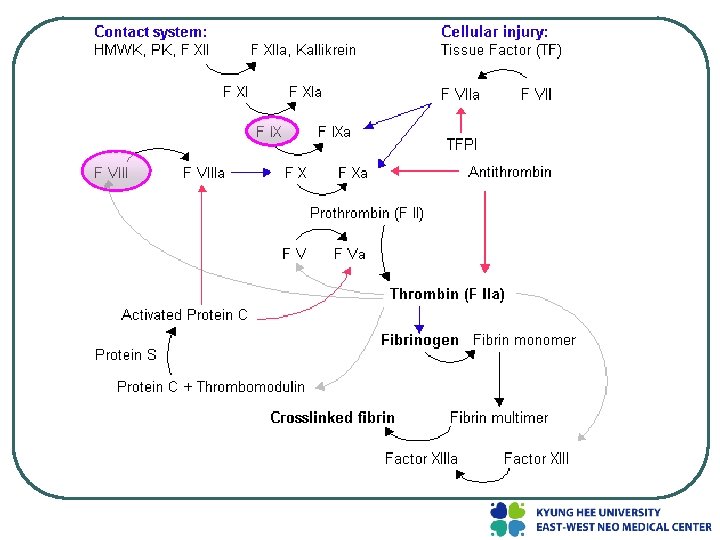
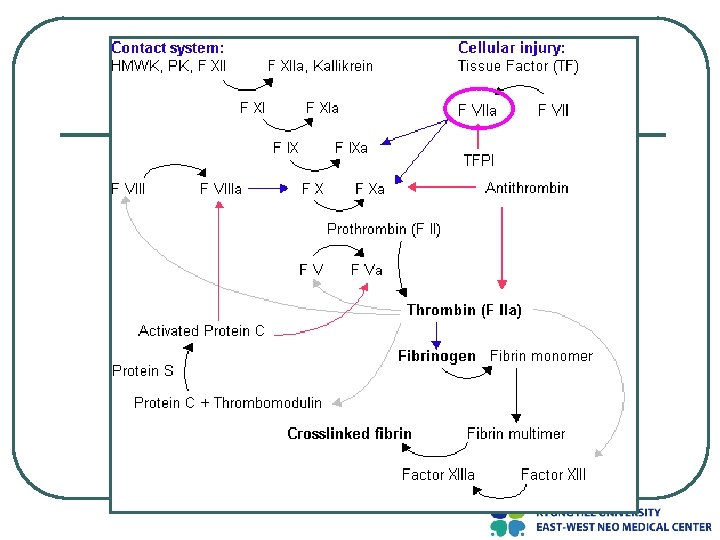
Classification l l l Hemophilia A - factor VIII deficiency, "classic haemophilia" (X-linked recessive) Hemophilia B - factor IX deficiency, "Christmas disease" (X-linked recessive) Hemophilia C - factor XI deficiency
l von Willebrand disease (v. WD) - milder than any of the three haemophilias - mutations in the coagulation protein von Willebrand factor. - the most common coagulation disorder present in 1% of the population.
Genetic structure
Clinical manifestations l Neither factor VIII nor factor IX crosses the placenta ; bleeding symptoms from birth or may occur in the fetus l Obvious symptoms of easy bruising, intramuscular hematomas, and hemarthroses l the hallmark of hemophilia is hemarthrosis l After repeated bleeding episodes into the same joint, patients with severe hemophilia may develop a "target" joint.
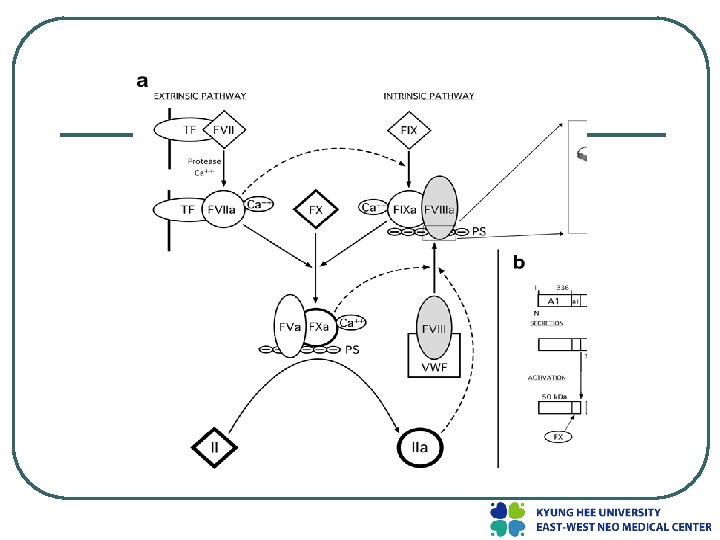
Diagnosis l l Hemoglobin/hematocrit Prothrombin time (PT) • • Extrinsic coagulation pathway screen Normal range expected Activated partial thromboplastin time (a. PTT) • • • Intrinsic pathway screen Elevated values expected May be normal range in mild disease Platelet count • • Assess bleeding Expect normal range
l l Factor VIII & IX level • • Assess percentage activity (normal 50 -150%). Expect severe disease with less than 1%, moderate disease with 1 -5%, and mild disease with greater than 5% Factor VIII & IX inhibitors • • • Assess presence. Assess anamnestic response to factor VIII. Expect low titer (0 -10 Bethesda U) or high titer (>10 Bethesda U)
Treatment l Basic Principles • should be treated with factor replacement therapy at the earliest possible moment, preferably within two hours of onset of symptoms. • Veins must be treated with care. • All products that cause platelet dysfunction ( ASA, NSAIDs ) ; with caution -> Paracetamol/acetaminophen • Avoid intramuscular injections. • Encourage home therapy with clotting factor concentrates.
l l l Severe hemophilia ; <1% activity of the specific clotting factor moderate hemophilia ; 1 -5% activity mild hemophilia ; >5% activity The hemostatic level for factor VIII is >30 -40% for factor IX, it is >25 -30%. The lower limit of levels for factors VIII and IX in normal individuals is approximately 50%.
Dose of FVIII (IU) = % desired (rise in FVIII) X body weight(kg) X 0. 5 Dise of FIX (IU) = % desired (rise in FIX) X body weight(kg) X 1. 4
Replacement therapy
Prophylaxis l lifelong prophylaxis to prevent spontaneous joint bleeding to maintain a measurable plasma level of clotting factor (1 -2 U/d. L)
Chronic complications l l l chronic joint destruction risk of transfusion-transmitted infectious diseases ( HIV infection, hepatitis C or B ) development of an inhibitor to either factor VIII or factor IX
1. Chronic arthropathy l Joint hemorrhage → proteolytic enzyme release macrophage proliferation ⇒ inflammation in the synovium ; synovium thickens frond-like projection into the joint
2. transfusion-transmitted infectious diseases l Fractionated plasma products ; a history of transmitting blood-borne viruses (HBV, HCV, and HIV) l Heat treatment ; generally effective against a broad range of viruses, both with and without a lipid envelope, including HIV, HAV, and HCV. l Solvent/detergent treatment ; effective against both HCV and HIV, but does not inactivate non-enveloped viruses, such as HAV.
3. inhibitor l About 10%-15% of hemophilia A patients and 1%3% of hemophilia B patients l early – within the first 10 -20 exposure days
Comprehensive care l best managed through comprehensive hemophilia care centers l Teams of physicians, nurses, orthopedists, physical therapists, and psychosocial workers
von Willebrand disease
v. WD l l 가성혈우병 the most common coagulation disorder present in 1% of the population autosomal inheritance menorrhagia ; major symptom
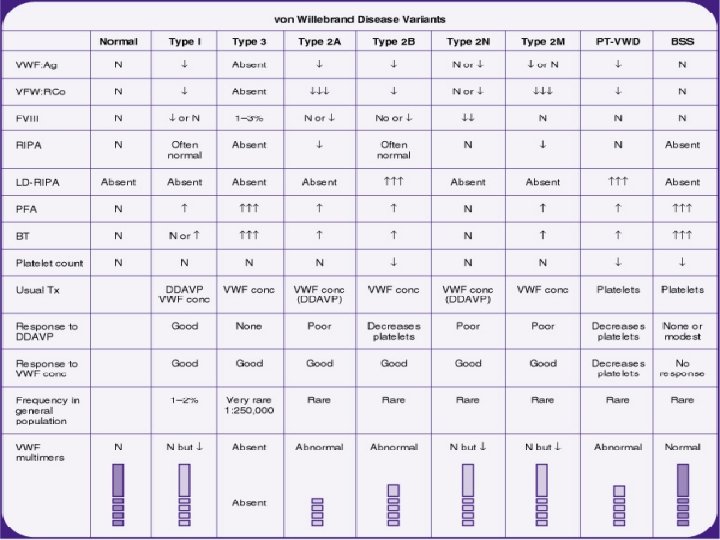
v. WD l classified into 3 main types, of which 70 -80% are considered to be type 1 • Type 1 ; partial quantitative decrease of qualitatively normal VWF and FVIII ; mild clinical symptoms ; autosomal dominant trait
v. WD • Type 2 ; 15 -20% have type 2 disease • • • ; variant of the disease with primarily qualitative defects of VWF Type 2 A VWD Type 2 B VWD Type 2 M VWD Type 2 N VWD Type 3 ; most severe form of VWD marked deficiencies of both VWF and FVIIIc the absence of VWF from both platelets and endothelial cells lack of response to DDAVP
v. WD Laboratory findings l long bleeding time and a long PTT frequently normal in patients with type 1 VWD quantitative assay for VWF antigen, VWF activity (ristocetin cofactor activity, or VWF: RCo), plasma factor VIII activity, determination of VWF structure (VWF multimers), and a platelet count
v. WD Treatment l Desmopressin • a synthetic analogue of antidiuretic hormone. • the primary treatment for bleeding • by causing release of von Willebrand factor (VWF) from endothelial storage sites l Greeneight® • Plasma-derived FVIII/v. WF
Other bleeding disorders
Factor VII deficiency Factor XI deficiency (Hemophilia C) Factor V deficiency Congenital afibrinogenemia Factor XIII defiency