HER 1EGFR and HER 2Erb B 2 pathways

HER 1/EGFR and HER 2/Erb. B 2 pathways O. Segatto, Regina Elena Cancer Institute

The hallmarks of cancer RTK activity regulates cellular programmes crucial to cell transformation and tumour progression

RTKs have intrinsic tyrosine kinase activity which is activated upon ligand binding Extracellular domain Kinase domain Ligand binding induces kinase activation and receptor self-phosphorylation on specific Tyr residues COOH tail RTKs are bistable systems, i. e. they transit from an “off” to an “on” state with no intermediate states

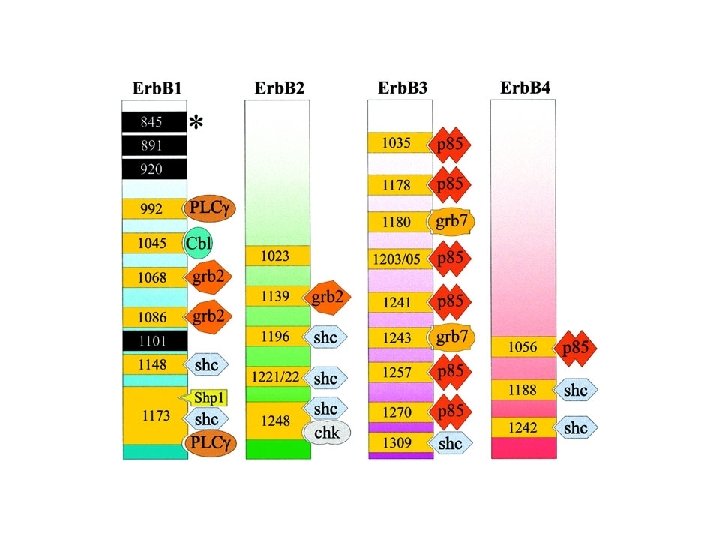
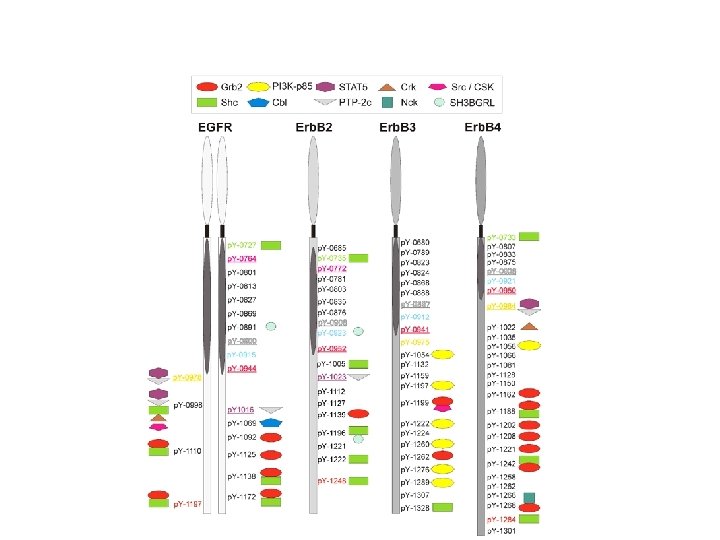
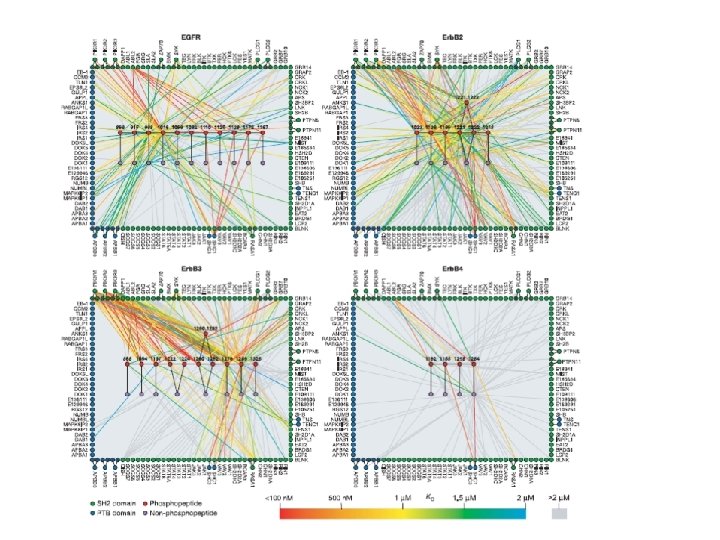
Specific p-Tyr sites generated by RTK autophosphorylation initiate downstream signalling by acting as docking sites for cellular proteins containing SH 2 or PTB domains. All SH 2 and some PTB domains bind to p-Tyr. Specificity of molecular recognition is dictated by residues C-terminal to p-Tyr for SH 2 domains and N-terminal to p-Tyr for PTB domains. The human genome encodes 139 SH 2 -containing and 49 PTB-containing proteins.

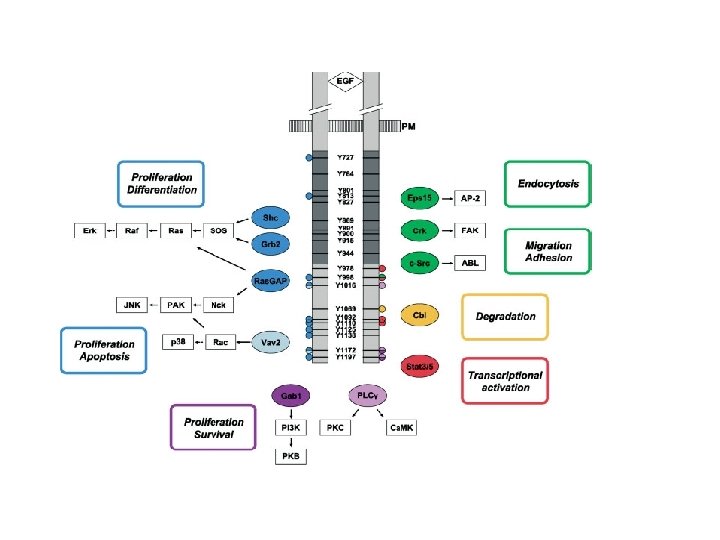
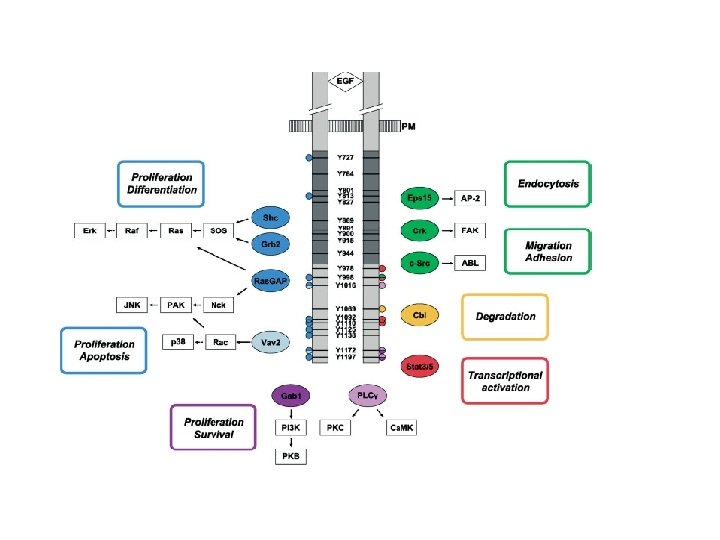
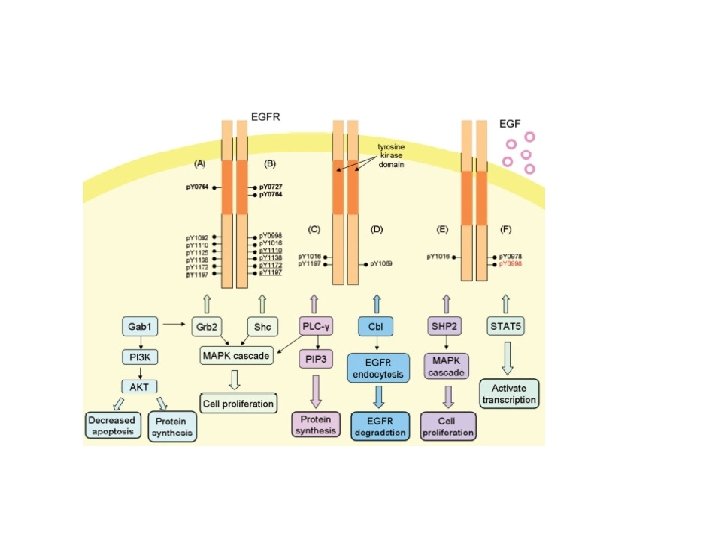
Activation of downstream signalling: enzymes containing an SH 2 domain bind to p. Y-EGFR and are relocated to the cell surface EGFR PIP 2 IP 3 + DAG PLC-g Relocation onto the EGFR allows PLC-g to be activated via Tyr phosphorilation and “induced proximity”

Activation of downstream signalling: SH 2 adaptors bind to p. Y-EGFR and relocate enzymes to the cell surface EGFR RAS RAF-ERK GRB 2 GTP (active) GDP (inactive) SOS PI-3 K-AKT

Direct plasmamembrane-to-nucleus signalling through engagement of STAT proteins

SH 2 and PTB domains are present in enzymes, adaptors and membrane-bound scaffolding proteins
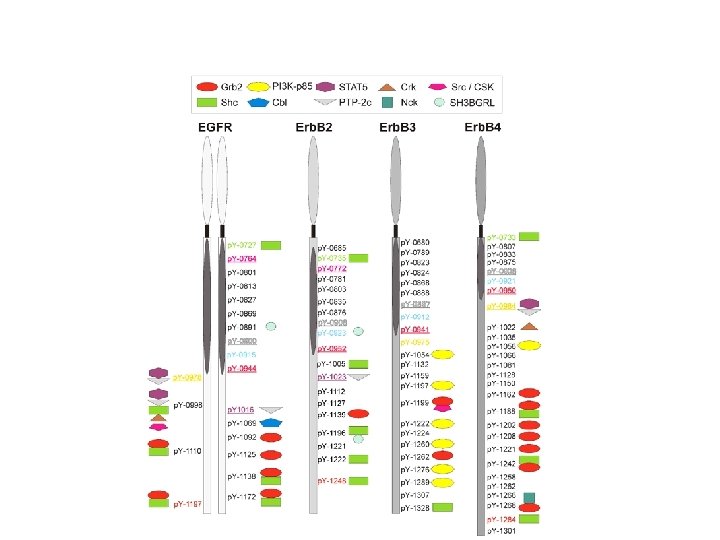

Formazione del “signalosoma” su RTK: perché? Concentrazione di effettori enzimatici in membrana Attivazione rapida e reversibile di funzioni enzimatiche Attivazione integrata di vie di segnalazione multiple Amplificazione del segnale Polarizzazione del segnale

RTK activation needs to be tightly controlled Aberrant RTK activity is linked to cell transformation

Focus Understanding the “design principles” of Erb. B activation, as deviation from these design principles may underline oncogenic conversion of Erb. B RTKs
 on-demand-only activation b) prevention of unwanted activation c) tight monitoring")
Key design principles a) on-demand-only activation b) prevention of unwanted activation c) tight monitoring of receptor activity Operational principles Intramolecular interactions lock receptors in an inactive conformation, which is released upon ligand binding Restricted ligand availability limits receptor activation Feedback inhibition restricts receptor activity

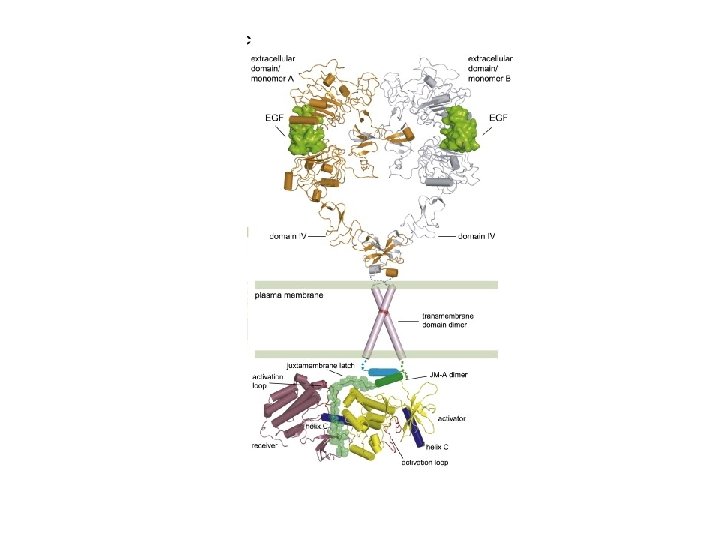
Intramolecular interactions lock EGF receptors in a monomeric inactive conformation, which is released upon ligand binding Part 1: structural transitions in the extracellular domain

Intramolecular interactions lock receptors in an inactive conformation, which is released upon ligand binding Part 2: structural transitions in the catalytic domain Xuewu Zhang et al. , Cell, Volume 125, Issue 6, 1137 -1149, 13 June 2006

Ligand-induced EGFR dimerization releases the intra-molecular inhibition of EGFR kinase via inter-molecular allosteric activation

Operational principles • Intramolecular interactions lock receptors in an inactive conformation, which is released upon ligand binding • Restricted ligand availability limits receptor activation • Feedback inhibition restricts receptor activity

Restricted ligand availability limits receptor activation Erb. B ligands are synthesised by stromal cells as membrane-bound precursors. Cleavage by proteases releases the soluble form

Operational principles • Intramolecular interactions lock receptors in an inactive conformation, which is released upon ligand binding • Restricted ligand availability limits receptor activation • Feedback inhibition restricts receptor activity

Feedback inhibition restricts EGF receptor activity P P PTP EGFR inhibitors X P P P

Feedback inhibition restricts receptor activity Downregulation depletes receptors and ligands, leading to cellular refractoriness to further homologous stimulation Inducible feedback regulators?

MIG 6 is an inducible feedback inhibitor that suppresses EGFR catalytic activation by binding to a dimer interface located in the COOH lobe of the EGFR kinase domain Xuewu Zhang, Kerry A. Pickin, Ron Bose, Natalia Jura, Philip A. Cole & John Kuriyan Nature 450, 741 -744(29 November 2007)

wt KO KO + Gefitinib

EGFR signalling activity is the result of a dynamic equilibrium between mechanisms of signal generation and signal extinction

RTKs are bistable systems, i. e. they transit from an “off” to an “on” state with no intermediate states Intramolecular interactions lock receptors in an inactive conformation, which is released upon ligand binding Restricted ligand availability limits receptor activation Feedback inhibition restricts receptor activity Oncogenic conversion of EGFR and ERBB 2 releases the receptors from these constrains, thus allowing unabated signalling activity

Erb. B receptors at work: the network context

Thomas Jefferson, United States Declaration of Independence, 1776 … whereas Erb. B receptors, ligands and ligand-receptor combinations are not!

Some ligands do not lead to EGFR degradation: TGFa and Epiregulin drive complete and fast EGFR recycling, Amphiregulin drives both fast and slow EGFR recycling. These ligands may be continuously re-used by the cell and also allow the EGFR to escape downregulation. High gain of signalling potency

Erb. B 2 and Erb. B 3 are non-authonomous receptors: Erb. B 2 does not bind to any known ligand, whereas Erb. B 3 has no kinase activity. Under physiological conditions Erb. B 2 and Erb. B 3 signal only in the context of ligand-induced heterodimers

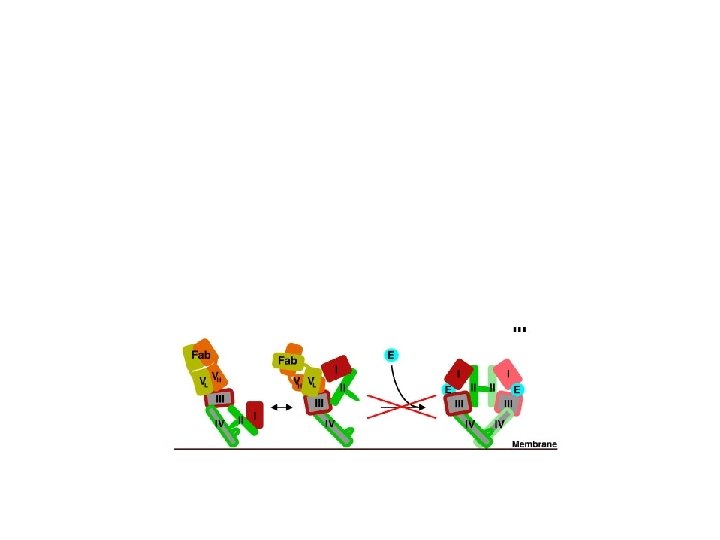
Erb. B 2/HER 2: an atypical RTK Sequence divergence in the extracellular region generates a) inability to bind ligand b) extended conformation of the dimerization arm Erb. B 2 is a powerful signal amplifier Erb. B 2 is the hierarchically dominant Erb. B receptor in dimer assembly Erb. B 2 is refractory to endocytosis/downregulation EGFR: Erb. B 2 heterodimers gain signalling potency due to decelerated ligand off-rates and refractoriness to endocytosis/downregulation

Strength and duration of signals generated by Erb. B RTKs depend on the nature of the ligand: dimer combination

Towards the system level… Citri et al. Nature Reviews Molecular Cell Biology 7, 505– 516 (July 2006)

Oncogenic conversion of EGFR and ERBB 2 in human tumours: mechanisms and therapeutic opportunities Oncogenic conversion is caused by genetic lesions, which drive the constitutive signalling activity of EGFR and ERBB 2. Oncogenic signalling by EGFR and ERBB 2 differs in quantitative and qualitative terms from physiological signalling. This may create a state of “oncogene addiction” and cause tumour cells to become exquisitively sensitive to drugs that target EGFR and/or ERBB 2.

EGFR mutations in lung cancer generate constitutively active kinases Inactive wt. EGFR L 858 R EGFR mutant Yun, C-H et al. , 2007, Cancer Cell, 11: 217 -227

Compound effects of mutational activation of EGFR in NSCLC Mutations cause constitutive activation of the EGFR kinase Mutations are associated to EGFR copy gain Mutations render EGFR refractory to down-regulation Mutations sensitize tumour cells to EGFR kinase inhibitors

ERBB 2 is activated by gene amplification and attendant over-expression – mutational activation is very rare Case study: breast cancer, ERBB 2 subtype ERBB 2 overexpression drives constitutive homo-dimerization ERBB 2 over-expression is associated to increased ERBB 3 expression, with ERBB 3 being a necessary signalling subunit of ERBB 2

RTKs are bistable systems, i. e. they transit from an “off” to an “on” state with no intermediate states Intramolecular interactions lock receptors in an inactive conformation, which is released upon ligand binding MUTATIONS, OVEREXPRESSION Restricted ligand availability limits receptor activation AUTOCRINE PRODUCTION of LIGANDS Feedback inhibition restricts receptor activity REFRACTORINESS to DOWN-REGULATION (several mechanisms) Oncogenic conversion of EGFR and ERBB 2 releases the receptors from these constrains, thus allowing unabated signalling activity
… … but also quality")
Quantity (strength and duration)… … but also quality

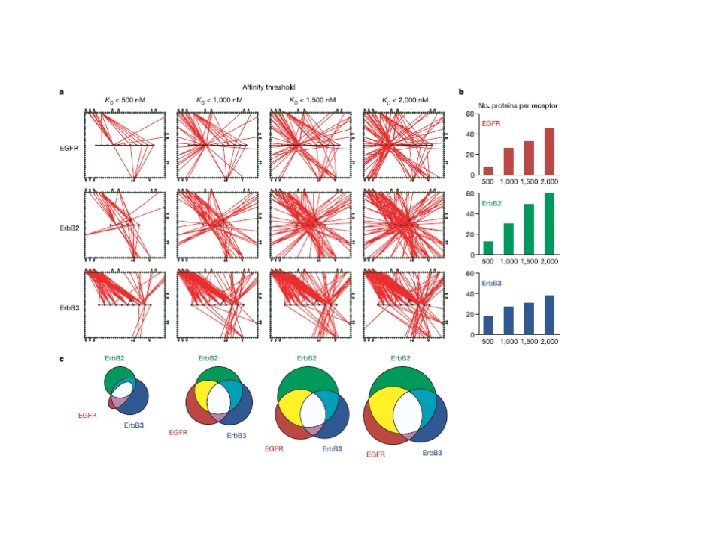
Signal promiscuity imposed by ERBB 2 average: 7. 2 average: 17 Jones, RB et al. , 2006 Nature, 439: 168 -174

Over-expression modifies the quality of signals generated by EGFR and ERBB 2 Jones, RB et al. , 2006 Nature, 439: 168 -174

Genetic lesions of EGFR and ERBB 2 in human tumours lead to constitutive signalling activity which differs from physiological signalling in quantitative and qualitative terms

Cetuximab inhibits ligand-dependent activation of EGFR

Targeting ERBB 2 oncogenic signalling with therapeutic antibodies

Kinase inhibitors target the ATP-binding pocket of EGFR and Erb. B 2

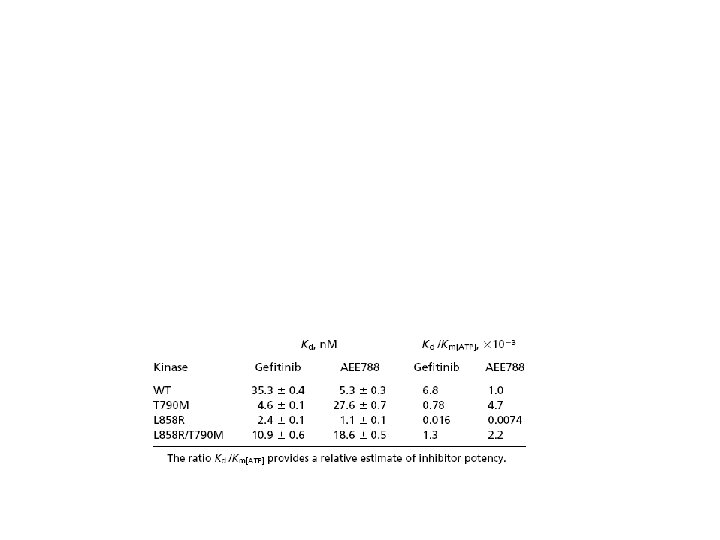
Mutations in the EGFR kinase may increase its affinity for competitive ATP inhibitors such as Gefitinib Different mutations may display different sensitivity to competitive ATP inhibitors: shall drugs get personal? Yun, C-H et al. , 2007, Cancer Cell, 11: 217 -227

Targeting the Erb. B network, rather than any individual Erb. B RTK may result in a therapeutic advantage Citri et al. Nature Reviews Molecular Cell Biology 7, 505– 516 (July 2006)

Conclusions Oncogenic signalling by EGFR and Erb. B 2 originates from the subversion of key regulatory principles of receptor activation Oncogenic conversion grants EGFR and Erb. B 2 full operational autonomy as well as evasion from negative regulation Targeting EGFR and Erb. B 2 in tumours must take into account the “design principles” of oncogenic signalling by EGFR and Erb. B 2, including aberrant network activation

In memoria di Matthias Kraus, 1956 -2009

X X
")
Citri et al. Nature Reviews Molecular Cell Biology 7, 505– 516 (July 2006)

Inactive wt. EGFR
- Slides: 61