Hemostasis Rafi Ahmed MD Hematology Oncology Fellow Hemostasis















 Xa (common")

 VIIa-TF")













 ▫ Deficiency in glycoprotein IIb and")





 • Thromboplastin (TF+PL+Ca 2+) added to citrated plasma • Trigger Extrinsic")



")


- Slides: 51
Hemostasis Rafi Ahmed, MD Hematology Oncology Fellow
Hemostasis • Process of blood clot formation at site of vessel injury • Response must be ▫ Quick ▫ Localized ▫ Carefully regulated
Phases of hemostasis • Formation of platelet plug • Propagation of clotting process by coagulation cascade • Termination of clotting by antithombotic control mechanisms • Removal of clot by fibrinolysis
Platelets • Production ▫ Produced in Bone Marrow by fragmentation of cytoplasm of megakaryocytes ▫ Differentiation of stem cell to production of platelets is approximately 10 days ▫ Thrombopoetin is regulator of platelet production which binds to C-MPL receptors on platelets ▫ Lifespan is 7 -10 days
Platelet Structure • Glycoproteins of the surface involved in platelet reactions of adhesion and aggregation • Interior with calcium, ADP, ATP, serotonin in granules. • Alpha granules with heparin antagonist, PDGF, Beta -thromboglobulin, fibrinogen, VWF, and clotting factors
Platelet Function • Formation of mechanical plugs during vascular injury ▫ ▫ Adhesion Aggregation Secretion Procoagulant activity
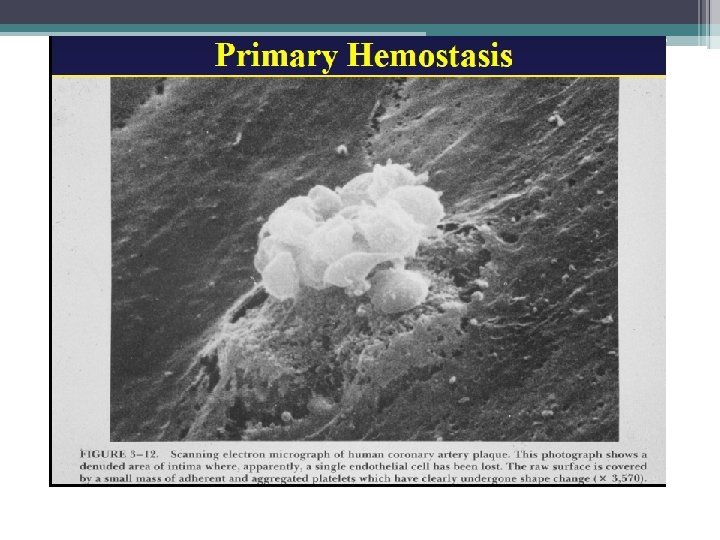
The Coagulation Cascade Primary Hemostasis • Consists of: ▫ Platelet plug formations ▫ Vascular spasm ▫ Capillary endothelial adhesions • Temporary Fix ▫ Lasts 12 -24 hours
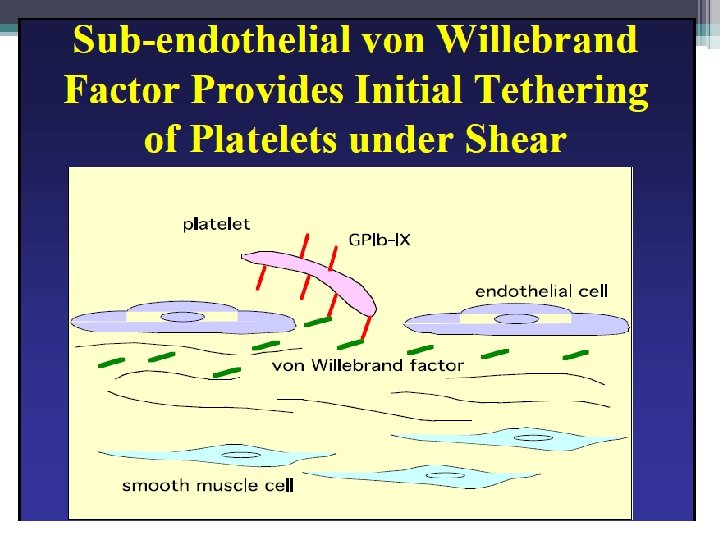
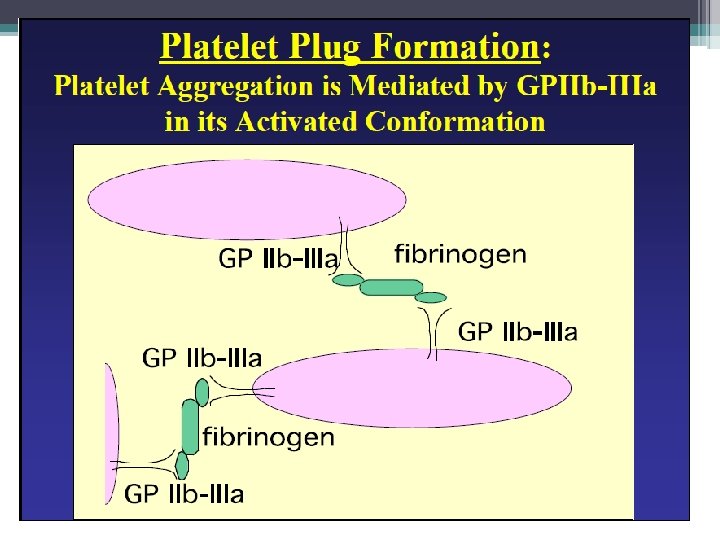
Primary Hemostasis • Platelets adhere to exposed endothelial tissue • Subendothelial microfibrils bind to subendothelial VWF (released from endothelium) This binds to GPIb-IX complex on platelet membrane • Platelets now “roll” along surface until GPIa/IIa binds to collagen • Platelet activation achieved by GPIIb/IIIa exposure to fibrinogen (primary) as well as VWF leading to platelet aggregation Hoffbrand et al. Essential Haematology. Oxford, UK: Blackwell Publishing, 2004.
Collagen Receptor GP IB/IIA GP IIB/IIIA
Primary Hemostasis • Platelets adhere to exposed endothelial tissue • Subendothelial microfibrils bind to subendothelial. VWF (released from endothelium) This binds to GPIb-IX complex on platelet membrane • Platelets now “roll” along surface until GPIa/IIa binds to collagen • Platelet activation achieved by GPIIb/IIIa exposure to fibrinogen (primary) as well as VWF leading to platelet aggregation Hoffbrand et al. Essential Haematology. Oxford, UK: Blackwell Publishing, 2004.
Primary Hemostasis: Platelet release reaction and aggregation • Collagen exposure/thrombin action: ▫ Secretion of platelet granules �ADP, serotonin, fibrinogen, lysosomal enzymes, B thromboglobulin, and heparin neutralizing factors • Activation of platelet prostaglandin synthesis ▫ Arachidonate release leads to thromboxane A 2 • Thromboxane A 2 ▫ Potentiates platelet aggregation ▫ Vasoconstrictive activity • Procoagulant Activity ▫ Phospholipid mediated reactions ▫ Factors IXa, VIIIa, and X in the formation of factor Xa ▫ Factors Xa, Va, and prothrombin II results in thrombin Hoffbrand et al. Essential Haematology. Oxford, UK: Blackwell Publishing, 2004.
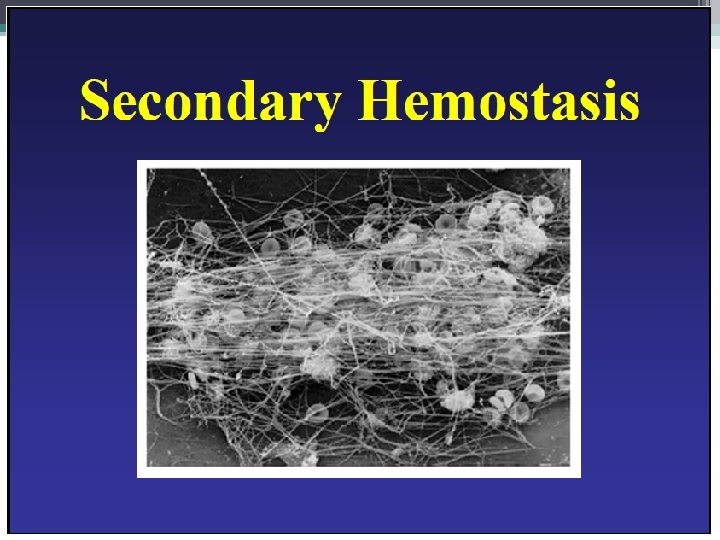
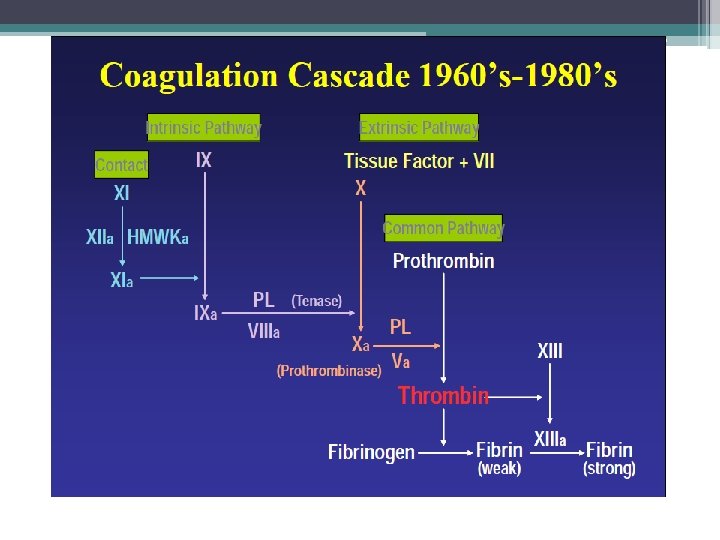
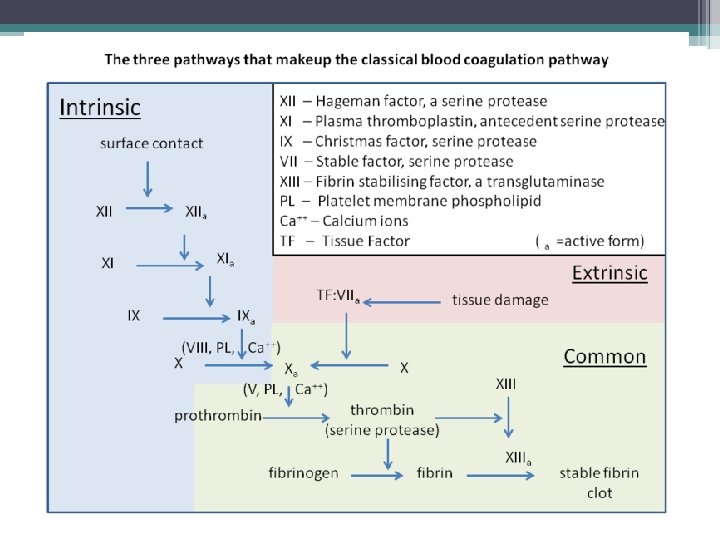
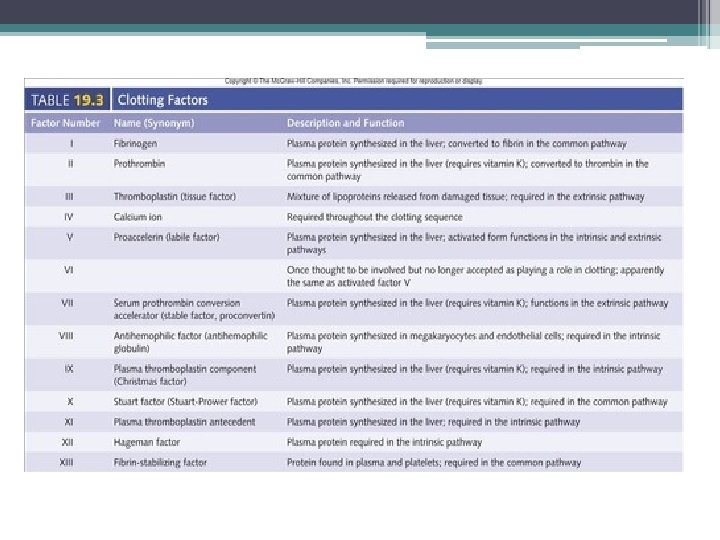
The Coagulation Cascade Secondary Hemostasis • Same time as platelets are aggregating • Initiation substances sequentially activate by proteolysis a cascade of circulating precursor proteins • Factors XII, XI, IX, VIII form the intrinsic pathway • VIIa-Tissue Factor complex form the extrinsic pathway • Both pathways leads to factor X activation • Final pathway is converting prothrombin to thrombin which converts fibrinogen to fibrin • Leads to firm and stable plugs
Secondary Hemostasis PTT PT Image retrieved from: http: //en. wikipedia. org/wiki/Coagulation
Cofactor
Tissue Factor • Cell bound glycoprotein • Principal initiator of coagulation in vivo • Mostly in extravascular location but gets exposed to blood when endothelial barrier is breached
Tissue Factor is a Co-Factor TF + VII X TF-VIIA (active enzyme) Xa (common pathway) IX IXa (intrinsic)
TFPI- Tissue Factor Pathway Inhibitor
In Vivo • TF binds to factor VII • Activates factor VII (VIIa) VIIa-TF complex • Activates factor IX and X • Factor X leads to small amounts of thrombin Thrombin • Activates cofactors V and VIII • Amplification involves VIII and IX • Continues to propagate factor Xa Activated factor X • Converts prothrombin to thrombin • Thrombin converts fibrinogen to fibrin monomer • XIIIa stabilizes fibrin polymers Secondary Hemostasis Hoffbrand et al. Essential Haematology. Oxford, UK: Blackwell Publishing, 2004.
Cofact or
Coagulation Factor Inhibitors • Important effect of thrombin is limited to site of injury • TFPI : tissue factor pathway inhibitor • Protein C and S: inhibitors of coagulation cofactor V and VIII ▫ Thrombin binds to endothelial surface receptor thrombomodulin ▫ Activates vitamin K dependent serine protease protein C ▫ Protein S binds protein C to the platelet surface • Anti Thrombin III
Protein C and Protein S Thrombomodulin/Thrombin Complex Thrombomodulin • Protein C-> Activated Protein C • Cell bound Protein on (enzyme) Endothelium • Protein S is cofactor for APC that binds • Binds to Thrombin (IIa) and it to platelet surface inactivates • Inactivates cofactors VIIIa and Va
Antithrombin III Antithrombin • Weak Inhibitor of enzymes until in activated confirmation • 1000 X fold activated in presence of heparin • Heparan sulfate proteoglycan on endothelial surface
Coagulation Factor Inhibitors • Fibrinolysis ▫ Generate Plasmin ▫ Remove Fibrin ▫ Fibrin �Cofactor for destruction
Fibrinolysis
Approach to Bleeding Patient • Abnormal Bleeding May Result From ▫ ▫ Vascular disorders Thrombocytopenia Defective platelet function Defective coagulation
Pattern of Bleeding • Vascular and Platelet Disorders ▫ Associated with bleeding from mucous membranes and into skin • Coagulation Disorders ▫ Bleeding in soft tissue or joints
Patient History • Bleeding History ▫ Past bleeding ▫ During surgical procedures ▫ Mucosal vs hemarthrosis • Medication Use ▫ Aspirin, clopidogrel, ticlopidine, warfarin
Inherited Vascular Disorders • Hereditary hemorrhagic telangiectasia ▫ ▫ Autosomal dominant Dilated microvascular swellings Pulmonary AV malformations Recurrent GI hemorrhage • Connective Tissue Disorders ▫ Ehlers-Danlos �Hereditary collagen abnormalities �Defective platelet aggregation
Acquired Vascular Defects • Senile purpura ▫ Atrophy of supporting tissues of cutaneous blood vessels on dorsal aspects of forearms and hands • Purpura due to infections ▫ Vascular damage from immune complex formation • Henoch-Schonlein ▫ In children, follows acute infection ▫ Ig. A mediated vasculitis • Scurvy ▫ Vitamin C deficiency, defective collagen • Steroid purpura ▫ Defective vascular support
Thrombocytopenia • Production Defects ▫ Bone Marrow Failure • Increased Destruction ▫ ▫ ▫ ITP HIT Drug induced TTP/HUS DIC Splenic Pooling
Hereditary Disorders of Platelets • Thrombasthenia (Glanzmann’s Disease) ▫ Deficiency in glycoprotein IIb and IIIa ▫ Platelets fail to aggregate • Bernard-Soulier Syndrome ▫ Platelets larger than normal ▫ Deficiency of glycoprotein Ib ▫ Defective binding to v. WF, adherence, and no aggregation in ristocetin ▫ Variable thrombocytopenia • Storage Pool Diseases
Acquired Platelet Functional Disorders • Liver disease ▫ Decrease in clotting factors ▫ Quantitative and qualitative platelet disorders ▫ Cirrhosis: sequestration, decreased TPO • Uremia ▫ Ecchymosis, epistaxis, GI/GU bleeding • Cardiopulmonary Bypass ▫ Platelet interaction with non-physiologic surfaces ▫ Hypothermia, complement activation, cytokines, thrombin ▫ Adhesive properties altered
Von Willebrands Disease • VWF Functions ▫ Primary Hemostasis �Platelet Adhesion �Platelet Aggregation ▫ Secondary Hemostasis �Carrier of VIII in plasma (keeps it inactivated) until thrombin releases it ▫ Synthesized in endothelial cells and megakaryocytes • Von Willebrand Disease ▫ Autosomal dominant
Von Willebrand Disease • Mucocutaneous bleeding usually mild to moderate • Assays ▫ VWF activity: measures VWF binding to plts �Ristocetin cofactor assay-gold standard �Collagen binding, VWF: activity assay ▫ VWF antigen: concentration of protein (elisa) ▫ Factor VIII activity (carrier function of VWF) ▫ VWF Multimers, RIPA (Assays for classification) • Type 1: Partial quantitative deficiency of VWF • Type 2: (A, B, M, N) Qualitative defect in VWF • Type 3: Total deficiency of VWF
Quick Review of Hemostasis Screening Assays • • Thrombin Time PT PTT Mixing Study
Thrombin Time • Simple test to measure conversion of fibrinogen to fibrin • Bovine thrombin added to citrated plasma, no calcium or phospholipid required (thus Lupus inhibitors don’t’ effect it) • Prolonged by ▫ ▫ ▫ Heparin Hypo or dysfibrinogenemia High levels of FDP Circulating Paraprotein Antibody to bovine thrombin (cardiac patients) • Reptilase time- will exclude Heparin ▫ Snake venom cleaves fibrinogen to fibrin ▫ Not affected by AT III
Prothrombin Time (PT) • Thromboplastin (TF+PL+Ca 2+) added to citrated plasma • Trigger Extrinsic Pathway • Usually insensitive to lupus inhibitors (since excess PL) • Usually insensitive to heparin (adding heparinase to reagent) • INR = (test PT/ mean normal PT) ^ISI (International sensitivity Index) Sensitivity to Deficiencies • Factors VII, X, V, II (30 -50 u/dl) (60 -160) • Fibrinogen 80 -100 mg/d. L (150 -400)
Activated Partial Thromboplastin Time APTT • Partial Thromboplastin= Phospholipid + Calcium • Activated: neg charged surface that activate contact factors (FXII, HMK) • Added to citrated Plasma with Calcium • Variable sensitivity to lupus inhibitors, heparin (less phospholipid)
Mixing Study • 50 -50 mix with normal plasma will correct if due to factor deficiency (50% of nl levels of any component factor) • If not correct then concern for inhibitor • Sometimes corrects but after incubation becomes prolonged- concern for inhibitor
Nl PT and prolonged PTT Disorders of intrinsic pathway Deficiencies • Factor VIII: Hemophilia A, VWD • Factor IX: Hemophilia B • Factor XI (variable bleeding history) • Factor XII, prekallikrein, HMWK (not associated with bleeding) Inhibitors • Factor VIII (most common) • Factor IX, XI (rare)
Prolonged PT and normal PTT • Deficiency ▫ Factor VII deficiency (range of phenotype) • Acquired inhibitors ▫ Factor VII rare • Most commonly seen in warfarin therapy, early liver disease, vitamin K deficiency, DIC
Prolonged PT and PTT • Deficiency of Factor X, V, II, I • Super therapeutic warfarin or heparin • Vitamin K deficiency, liver disease, DIC, fibrinolysis
And finally some anticoagulants • Vitamin K antagonists ▫ Warfarin (factor 10, 9, 7, 2, C, S) • Activates Antithrombin III ▫ Heparin, LMWH ▫ Fondaparinux (pentasacchride) • Direct Xa Inhibitors ▫ Rivaroxaban, apixaban • Direct Thrombin Inhibitors ▫ Argatroban, Dabigatran