Hemostasis and Coagulation Mikls Molnr Definition of HEMOSTASIS




 platelet exposure to subendothelial connective tissue of blood vessels")




 glycocalyx, plasma membrane Sol-gel zone")
 Receptor site for v. WF IIb, IIIa (GPIIb/IIIa) Complex becomes")

 Hemostatic proteins Fibrinogen Factor V v. WF Plasminogen activator inhibitor")






 Fibrinogen group: I, V, VIII, XIII most labile, are consumed")
 Tissue")







 α 2 –antiplasmin α 2 -macroglobulin")





 Connective tissue disorders")

")
















 Acute - children (post infection) Chronic - adults ( females,")


")
 X-linked disorder (affects 1º males) Prevalence is")
 X-linked recessive disorder Prevalence is 1: 30,")
 Inherited form transmitted as an autosomal recessive trait. Prevalence")

 Major pathologic processes obstetric")


- Slides: 67
Hemostasis and Coagulation Miklós Molnár
Definition of HEMOSTASIS The arrest of bleeding by repair of vessel wall Maintaining a balance Coagulation Fibrinolysis Hypocoagulation: excessive bleeding (inherited or acquired) Hypercoagulation (thrombosis) inadequate activation of the fibrinolytic system
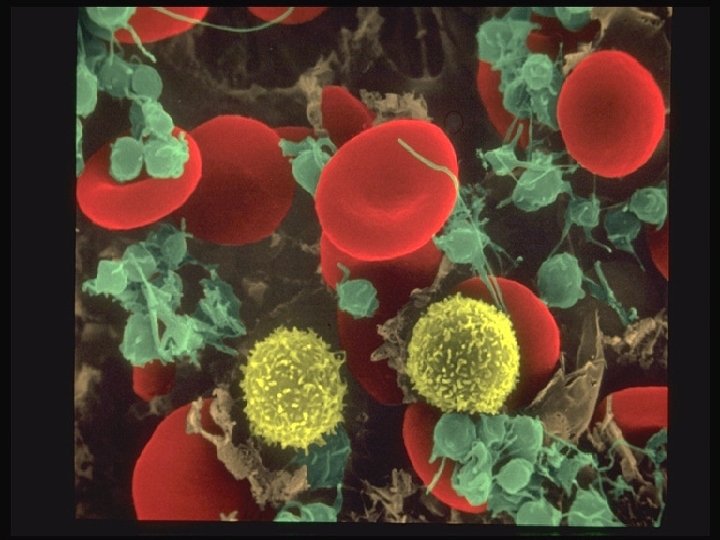
Systems Involved in Hemostasis Vascular system Injured vessel initiates vasoconstriction Platelet System Injured vessel exposes collagen that initiates platelet aggregation and help form plug Coagulation System protein factors of intrinsic and extrinsic pathways produce a permanent fibrin plug
HEMOSTASIS Primary vs. Secondary vs. Tertiary Primary Hemostasis Platelet Plug Formation Dependent on normal platelet number & function Initial Manifestation of Clot Formation Secondary Hemostasis Activation of Clotting Cascade Deposition & Stabilization of Fibrin Tertiary Hemostasis Dissolution of Fibrin Clot Dependent on Plasminogen Activation
Primary Hemostasis vasoconstriction (vascular system) platelet exposure to subendothelial connective tissue of blood vessels Platelet release of ADP, ATP, Thromboxane A 2 (promotes vasoconstriction) Platelet aggregation, phospholipid provides site for fibrin formation
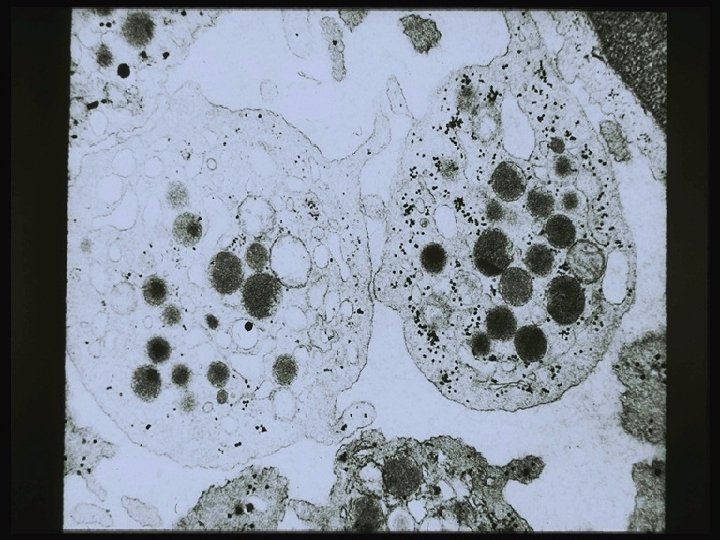
Platelet Origin & Development Endomitosis Multiple mitotic division of DNA without cytoplasmic division Largest cell in the BM low power Stem cell to mature platelet = 5 days Each megakaryocyte can shed 500 -4000 platelets Cytoplasm breaks apart along demarcation membranes
Megakaryocyte
Platelets forming from cytoplasm
Normal platelets and one giant platelet
Platelet Structure Three zones Peripheral zone (adhesion & aggregation) glycocalyx, plasma membrane Sol-gel zone (structure & support) microfilaments, thrombosthenin, open canalicular system, dense tubular system Organelle zone (Secretion & storage) granules: alpha, dense, glycogen mitochondria, lysosomes
Glycocalyx Glycoproteins Ib (GPIb) Receptor site for v. WF IIb, IIIa (GPIIb/IIIa) Complex becomes receptor site for fibrinogen
Granular content Dense granules ATP ADP Calcium Magnesium Serotonin epinephrine
Granular content (Alpha granules) Hemostatic proteins Fibrinogen Factor V v. WF Plasminogen activator inhibitor (PAI-1) α 2 -antiplasmin Nonhemostatic proteins β-thromboglobulin, Platelet factor 4 Platelet derived growth factor (PDGF) Albumin fibronectin,
Formation of primary hemostatic plug Platelets converted from inactive to active state Adhesion to collagen Triggers platelet activation Tromboxane A 2 is synthesized from arachidonic acid and stimulates secretion Aggregation of platelets to each other prostacyclin (PGI 2) inhibits platelet aggregation Secretion (discharge of granule contents)
VON WILLEBRAND FACTOR Large Adhesive Glycoprotein Polypeptide chain: 220, 000 MW Base structure: Dimer; Can have as many as 20 linked dimers Multimers linked by disulfide bridges Synthesized in endothelial cells & megakaryocytes Constitutive & stimulated secretion Large multimers stored in Weibel-Palade bodies Functions: 1) Stabilizes Factor VIII 2) Essential for platelet adhesion
Stable adhesion Platelet activation/ Platelet aggregation Rolling Blood Flow VWF Platelet adhesion VWF collagen VWF
Inactive Active
Secondary hemostasis Intrinsic Pathway All components required for initiating this pathway are circulating in the blood triggered by contact with collagen or glass Extrinsic Pathway Initiated by the release of tissue thromboplastin and calcium from damaged tissue Common Pathway Leads to clot formation including the platelet plug and fibrin produced
Coagulation Proteins Zymogens enzyme precursors II, VII, IX, X, XII, Prekallkrein When activated become serine proteases Cofactors Nonenzymatic V, VIII, HMWK, Tissue factor(thromboplastin) Kinin factors prekallikrein, HMWK Roles include coag activation as well as fibrinolytic activation
Coag factors (by group) Fibrinogen group: I, V, VIII, XIII most labile, are consumed in coagulation, found on platelets Prothrombin group: II, VII, IX, X Vitamin K dependent, may be affected by coumarin, diet, antibiotics Contact group: XI, XII, HMWK, Prekallikrein initiate intrinsic path and fibrinolysis
VIIa + Tissue factor pathway inhibitor X Positive feedback Xa Tissue Factor (TF) Tissue Damage Vessel wall Cell particles II IIa (prothrombin) (thrombin) Initial Tissue Factor Pathway Activation of Hemostasis
IIa Thrombin Pro-coagulant effects XI XIa VIIIa V Fibrinogen Va Fibrin
XIa IX Precursor IXa Enzyme Reaction on Activated Platelets FVIIIa/Ca 2+/Phospholipid X Xa FVa/Ca 2+/Phospholipid II Fibrinogen IIa Fibrin
FIBRINOLYTIC SYSTEM Definition: temporary fibrin clot systematically and gradually dissolved as the vessel heals Key components Plasminogen (inactive form) Plasminogen activators Plasmin Fibrin Degradation Products (FDP) Inhibitors of plasminogen activators and plasmin
Activators of Fibrinolysis Intrinsic activators Factor XIIa, XIa, kallikrein Extrinsic activators Tissue type plasminogen activator (t-PA) Urokinase type plasminogen acitvator (u-PA) Exogenous activators Streptokinase (derived from beta strep)
XII XIIa XI Protein S XIa IX IXa Protein S Protein C VIIIa+Ca+Pl X Xa Va+Ca+Pl TF / VIIa TFPI II Fibrinogen IIa/Thrombomodulin interaction IIa Fibrinolysis
VIIIa Va Protein S IIa Thrombin Anti-coagulant effects Activated Protein C IIa thrombomodulin Protein C Anticoagulant Pathway
Coagulation Regulatory Mechanisms Naturally Occurring Anticoagulants rapidly interact with components of coag cascade to avoid unabated clot formation Protein C (PC) and Protein S (PS) deficiencies may be congenital or acquired Antithrombin (AT) and Heparin Cofactor II serine protease inhibitors (serpins) Deficiency of inhibitors cause increased risk of thrombosis
Inhibitors of Fibrinolysis Plasminogen Activator Inhibitors (PAI) α 2 –antiplasmin α 2 -macroglobulin
Bleeding disorders
Disorders of Hemostasis Vascular disorders Scurvy, easy bruising, Henoch-Schönlein purpura. Platelet disorders Quantitative - Thrombocytopenia Qualitative - Platelet function disorders – Glanzmans Coagulation disorders Congenital - Haemophilia (A, B), Von-Willebrands Acquired - Vitamin-K deficiency, Liver disease Mixed/Consumption: DIC
Tests of Hemostasis: Screening tests: Bleeding. T 10 m. Platelet & BV function Prothrombin. T Extrinsic, a. PTT Instrinsic Thrombin. T common path. (DIC) Specific tests: Factor assays – hemophilia. Tests of thrombosis – TT, FDP, DDA, Platelet function studies: Adhesion, Aggregation, Release tests. Bone Marrow study
Bleeding: Clinical Features Local-vs- General, spontaneous… Hematoma / Joint Bleeds- Coag Skin / Mucosal Bleeds – PLT wound / surgical bleeding – Immediate - PLT Delayed - Coagulation
Platelet Coagulation Petechiae, Purpura Hematoma, Joint bl.
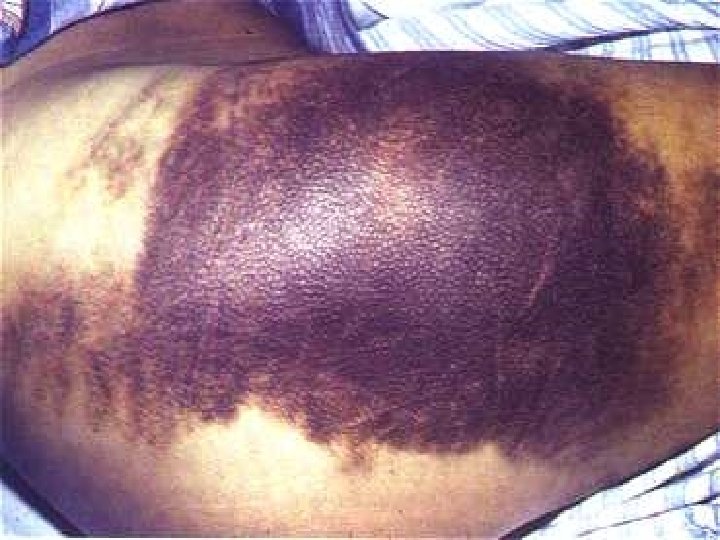
Vascular disorders: Petechiae, purpura, ecchymoses senile purpura vitamin C deficiency (scurvy) Connective tissue disorders Infections – Meningococcus Henoch-Schonlein Purpura-Immu
Senile Purpura
Petechiae in Vasculitis (Rocky Mountain Spotted Fever)
Henoch-Schölein purpura Immune disorder Children Follows infection Petechiae with edema and itching.
Henoch-Schönlein purpura 20 y Male, fever, painful symmetric polyarthritis for a day. During the next two days, edema and palpable purpura developed.
Platelet Disorders - Features: Mucocutaneous bleeding Petechiae, Purpura, Ecchymosis. Spontaneous bleeding after trauma CNS bleeding (severe plt) Prolonged bleeding time (BT)
BLEEDING TIME vs. PLATELET COUNT
REDUCED PLATELET NUMBER: THROMBOCYTOPENIA Normal platelet count 140 -400 Increased bleeding time <100 Spontaneous bleeding <20
PLATELET FUNCTION DEFECTS Prolonged Bleeding Time Congenital Drugs Alcohol Uremia Hyperglobulinemias Fibrin/fibrinogen split products Thrombocythemia Cardiac Surgery
PLATELET FUNCTION DEFECTS Platelet Adhesion Bernard Soulier Disease Abnormal GPIb-IX Complex Receptor for von Willebrand factor Only adhesion mediator @ high shear stress Tested by ability to aggregate platelets in presence of ristocetin Von Willebrand disease Reduced or dysfunctional von Willebrand factor
Von-Willebrand Disease: Coagulation + PLT disorder: Congenital disorder Deficiency of v. WF molecule Part of FVIII, Mediates platelet adhesion Prolonged Bleeding time Low Factor VIII & long a. PTT Mucocutaneous bleeding
Von-Willebrand Disease v. WF: F-VIII & PLT function. Defective Platelet Adhesion Skin Bleeding Prolonged Bleeding time. Low Factor VIII levels. Prevalence: 0. 8– 2% (probable underestimate)
PLATELET FUNCTION DEFECTS Platelet Release Defects - Congenital -storage pool disease Failure to form dense granules syndrome) (Hermansky-Pudlak Do not release ADP, serotonin, calcium on activation Fail to recruit platelets for aggregation Gray platelet syndrome Failure of packaging of α-granules Do not release protein mediators of platelet aggregation Decreased platelet aggregation Mild bleeding disorder
PLATELET FUNCTION DEFECTS Aggregation-Congenital Glanzmann's thrombasthenia Autosomal recessive Lack of fibrinogen receptor, GP IIb/IIIa Platelets cannot aggregate in response to usual stimuli Bleeding sometimes severe
Platelet Aggregation Curves
PLATELET FUNCTION DEFECTS Acquired - Drug Induced Alcohol Prostaglandin Synthetase Inhibitors Aspirin Non-Steroidal Antiinflammatory Drugs Phenylbutazone ADP receptor inhibitors Clopidogrel Ticlopidine Beta-lactam antibiotics Heparin
THROMBOCYTOPENIA Decreased production Decreased megakaryocytes Normal platelet life span Good response to platelet transfusion Neoplastic Causes Leukemias Aplastic Anemia Metastatic Carcinoma Drugs Radiotherapy Primary Marrow Disorders Megaloblastic Anemias Myelodysplastic syndromes Myeloproliferative diseases Some congenital syndromes
THROMBOCYTOPENIA Increased Destruction Shortened platelet life span Increased megakaryocytes Macroplatelets Poor response to platelet transfusion
THROMBOCYTOPENIA Increased Destruction - Causes Immune ITP Lymphoma Lupus/rheumatic diseases Drugs Consumption Disseminated intravascular coagulation Thrombotic thrombocytopenic purpura Hemolytic/uremic syndrome Septicemia
IDIOPATHIC THROMBOCYTOPENIA PURPURA (ITP) Acute - children (post infection) Chronic - adults ( females, 20 -40 yrs) Ig. G autoantibodies bound to platelets Platelets removed by macrophages Antibodies can act on marrow No good diagnostic test Treatment - Inhibit macrophage clearance Corticosteroids High dose gamma globulin Splenectomy
HIV-ASSOCIATED THROMBOCYTOPENIA Early Immune mediated Often in absence of AIDS Remainder of marrow WNL Treatment - Antiretroviral therapy Late Usually marrow infiltration Often pancytopenia Often associated infection or neoplasm Poorly responsive to all treatments
Coagulation disorders: Deficiencies of Clotting factors Onset - delayed after trauma Deep bleeding Into joints - Hemarthroses Into deep tissues – Hematoma large skin bleed – Ecchymoses
Coagulation Disorders Laboratory findings: Normal bleeding time & Platelet count Prolonged prothrombin time (PT) deficiencies of II, V, VII, X Prolonged time (a. PTT) all factors except VII, XIII Mixing studies - normal plasma corrects PT or a. PTT
Factor VIII Deficiency Classic hemophilia (Hemophilia A) X-linked disorder (affects 1º males) Prevalence is 1: 5, 000 males Most common - severe bleeding Spontaneous hematomas Abnormal a. PTT – Intrinsic path. Diagnosis - factor VIII assay Treatment - factor VIII concentrate Cryoprecipitate (less desirable)
Factor IX Deficiency Christmas disease (Hemophilia B) X-linked recessive disorder Prevalence is 1: 30, 000 males Indistinguishable from classic hemophilia (F VIII) Requires evaluation of factor VIII and IX activity levels to diagnose Treatment - factor IX concentrate Cryoprecipitate if factor IX unavailable
FACTOR XI DEFICENCY (Hemophilia C) Inherited form transmitted as an autosomal recessive trait. Prevalence is 1: 100, 000 Increased prevalence in Ashkenazi Jewish population (in Israel, estimated at 8%) A clinically mild bleeding problem Prolongs only the PTT Most often associated with liver disease
Secondary Hemostatic Disorders Vitamin K deficiency Neonates - decreased intestinal flora and dietary intake Oral anticoagulants (coumadin) Fat malabsorption syndromes Required for factors II, VII, IX, - Prolonged PT and a. PTT
Combined Primary and Secondary Hemostatic Disorders Disseminated Intravascular Coagulation (DIC) Major pathologic processes obstetric complications, neoplasms, infection (sepsis), major trauma Primary - platelet consumption ( bleeding time, platelets) Secondary - factor consumption ( PT, a. PTT)
Combined Primary and Secondary Hemostatic Disorders Severe Liver Disease Primary - dysfunctional platelets and/or thrombocytopenia ( BT) Secondary - decrease in all coagulation factors except v. WF ( PT, a. PTT) Vitamin K will promote synthesis of factors II, VII, IX, X
Summary