HEMOLYTIC ANEMIAS RED CELL MEMBRANE AND METABOLIC DEFECTS

HEMOLYTIC ANEMIAS: RED CELL MEMBRANE AND METABOLIC DEFECTS Normocytic normochromic anemia

NORMAL RED CELL MEMBRANE Structure • The detailed architecture of the red cell membrane is complex, but its basic design is relatively simple. • The lipid bilayer incorporates phospholipids and cholesterol, and it is spanned by a number of proteins that have their hydrophobic transmembrane domains embedded in the membrane. • Most of these proteins have hydrophilic domains extending toward both the outside and the inside of the cell. • Other proteins are tethered to the membrane through a glycosylphosphatidylinositol (GPI) anchor, and they have only an extracellular domain. • These proteins are arranged roughly perpendicular to or lying across the membrane: they include ion channels, receptors for complement components, receptors for other ligands, and some of unknown function.

• The most abundant of these proteins are glycophorins and the so-called band 3, an anion transporter. • The extracellular domains of many of these proteins are heavily glycosylated, and they carry antigenic determinants that correspond to blood groups. • Underneath the membrane, and tangential to it, is a network of other proteins that make up the cytoskeleton: • the main cytoskeletal protein is spectrin, the basic unit of which is a dimer of α-spectrin and β-spectrin. • The membrane is physically linked to the cytoskeleton by a third set of proteins (including ankyrin and the so-called band 4. 1 and band 4. 2), which thus make these two structures intimately connected to each other.
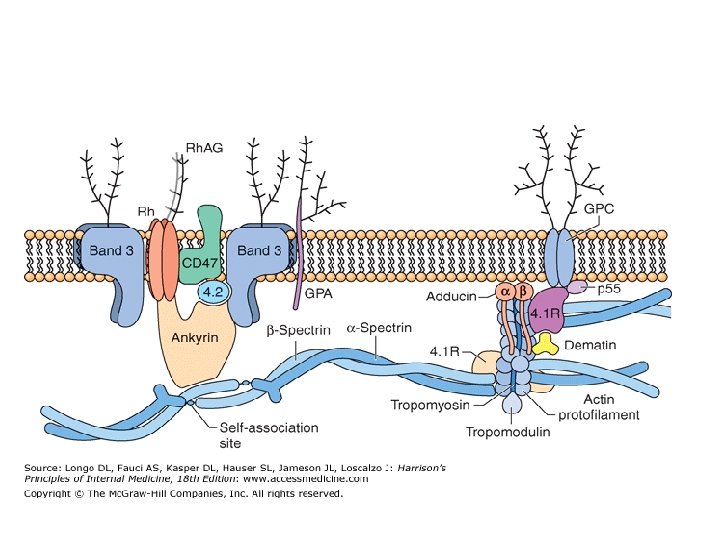

• The membrane-cytoskeleton complex is indeed so integrated that, not surprisingly, an abnormality of almost any of its components will be disturbing or disruptive, causing structural failure, which results ultimately in hemolysis. • These abnormalities are almost invariably inherited mutations; thus, diseases of the membranecytoskeleton complex belong to the category of inherited HAs. • Before the red cells lyse, they often exhibit more or less specific morphologic changes that alter the normal biconcave disk shape. • Thus, the majority of the diseases in this group have been known for over a century as hereditary spherocytosis and hereditary elliptocytosis.
 mechanical coupling")
membrane Function • Membrane Strength and Deformability : • The exquisite (perfect) mechanical coupling between the membrane skeleton and the overlying lipid bilayer confers on the normal red cell its remarkable properties of strength and deformability. • On release from the bone marrow, mature red cells must withstand the high pressure and shear forces in the heart and large arteries and also traverse the small-diameter microcirculatory vessels for 120 days. • The ability of the skeleton-bilayer couple to withstand high shear and to deform readily allows normal red cells to perform these tasks. • Abnormal red cells with defective membrane skeletons or defective coupling between the skeleton and the overlying bilayer fragment spontaneously in the circulation, which leads to the clinical picture of non-immune hemolytic anemia.

• • • Cation and Volume Homeostasis The red cell uses protein pumps and channels in its lipid bilayer membrane to control intracellular concentrations of sodium, potassium, and calcium ions and thereby to regulate cell volume. Normal intracellular concentrations of Na+, K+, and Ca 2+ are about 10 mmol/L, 100 mmol/L, and 100 nmol/L, respectively. The physiologically most important pumps include the ATP-dependent Na+-K+ exchanger, which uses the energy provided by ATP hydrolysis to pump Na+ outward against its concentration gradient and K+ inward against its concentration gradient, and the ATP-dependent Ca 2+ pump, which pumps Ca 2+ outward against its concentration gradient. The activities of these pumps counteract the small passive leaks of Na+, K+, and Ca 2+ down their concentration gradients through the relatively impermeable lipid bilayer. Pathologic increases in the passive leak rates of these three cations—or decreases in the activities of these two pumps—can have deleterious effects. A net gain of intracellular cations obligates net water entry and causes cells to swell, whereas a net loss of intracellular cations dehydrates cells. The free flow of water molecules in both directions across the lipid bilayer is mediated by the aquaporin-1 water channel protein. A pathologic increase in intracellular Ca 2+ concentration can be especially harmful by (1) activating a Ca 2+-dependent K+ channel (the Gardos channel) that mediates K+ efflux and cell dehydration and (2) at very high concentrations, activating a Ca 2+-dependent transglutaminase that cross-links membrane proteins and thereby (among other effects) decreases cell deformability.

• Cell Shape • The biconcave disc shape of normal red cells is maintained by a balance of forces within the membrane skeleton and between the skeleton and the lipid bilayer. • These forces are sufficiently robust to allow normal red cells to deform without fragmenting in the normal circulation. • Alterations in membrane skeleton integrity, skeleton-bilayer coupling, intracellular cation and water content, transmembrane protein organization, and hemoglobin denaturation and polymerization can affect red cell morphology. • One major determinant of red cell shape is the ratio between the surface area and volume of the cell; decreases and increases in this ratio lead to the formation of sphere-shaped (spherocyte) and cup(stomatocyte) or target-shaped red cells, respectively. • Irreversible shape change can also be mediated by permanent deformation of the membrane skeleton; orderly plastic deformation causes the formation of elliptical or oval red cells (elliptocytes or ovalocytes), whereas random membrane injury with denatured hemoglobin precipitation on the skeleton and oxidative cross-linking of proteins leads to the formation of spiculated (echinocyte), irreversibly sickled, and other abnormal red cell forms.

• Anion Exchange • The red cell membrane plays an important physiologic role in carbon dioxide (CO 2) transport. • CO 2 handling is facilitated by red cell enzyme-mediated conversion of this molecule to bicarbonate (HCO 3 -) in the tissues and back to CO 2 for excretion in the lungs. • To increase the HCO 3 --carrying capacity of the blood, some of the HCO 3 - is carried in the plasma. Movement of HCO 3 - in and out of red cells is facilitated by the presence of about 1 million anion exchanger molecules (band 3 proteins) in each red cell membrane. • Band 3 mediates the passive bidirectional exchange of HCO 3 - for Cl-; no energy is required for this process. Band 3 therefore serves at least two important roles in red cell membrane structure and function: coupling the membrane skeleton to the overlying lipid bilayer and mediating anion exchange across the membrane.

• Interactions with the Spleen: Red Cell Senescence • Most normal red cells are removed from the circulation by the spleen after a 120 -day lifespan. • The fenestrations between splenic cords and sinuses provide mechanical stress as red cells squeeze through these openings, whereas the lowoxygen, low-glucose, low-p. H environment of the splenic cords places metabolic stress on the cells. • The spleen uses two major mechanisms to sequester and remove aged red cells: • First, as red cells become less deformable with age, they are less able to traverse the splenic fenestrations. Second, as red cells age, their membranes are progressively decorated with autoantibodies or complement proteins that bind to receptors on mononuclear phagocytes in the spleen; these autoantibodies may be directed against clustered or proteolytically altered band 3 at the red cell surface.
 • Autosomal dominant common northern European people, although the disease can")
Hereditary Spherocytosis (HS) • Autosomal dominant common northern European people, although the disease can occur in any population. • This is a relatively common type of HA, with an estimated frequency of at least 1 in 5000. • HS came to be defined as an inherited form of HA associated with the presence of spherocytes in the peripheral blood. • In addition the red cells were abnormally susceptible to lysis in hypotonic media: indeed, the presence of osmotic fragility became the main diagnostic test for HS. • Today we know that HS, thus defined, is genetically heterogeneous; i. e. , it can arise from a variety of mutations in one of several genes
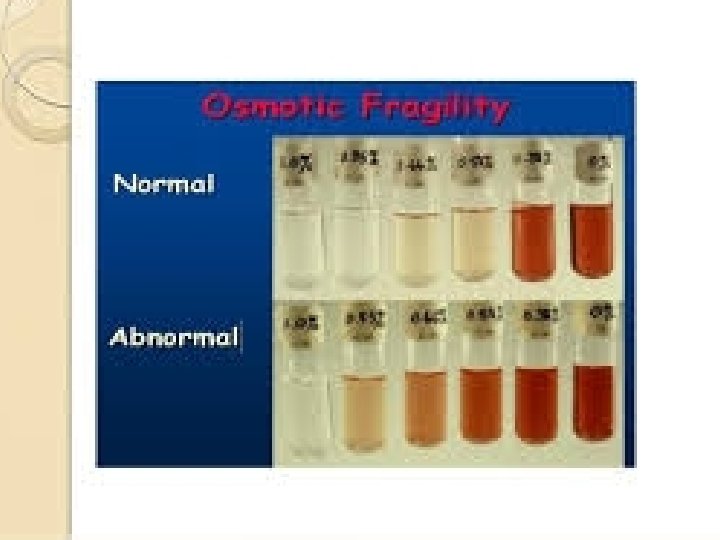

Clinical Presentation and Diagnosis • The spectrum of clinical severity of HS is broad. • Severe cases may present in infancy with severe anemia, whereas mild cases may present in young adults or even later in life. • In women, HS is sometimes first diagnosed when anemia is investigated during pregnancy. • The main clinical findings are jaundice, an enlarged spleen, and often gallstones; indeed, it is often the finding of gallstones in a young person that triggers diagnostic investigations. • In milder cases hemolysis is often compensated, and this may cause variation in time, even in the same patient, due to the fact that intercurrent conditions (e. g. , infection) cause decompensation. • The anemia is usually normocytic, with the characteristic morphology that gives the disease its name. • A characteristic feature is an increase in mean corpuscular hemoglobin concentration (MCHC): this is almost the only condition in which an increased MCHC is seen.
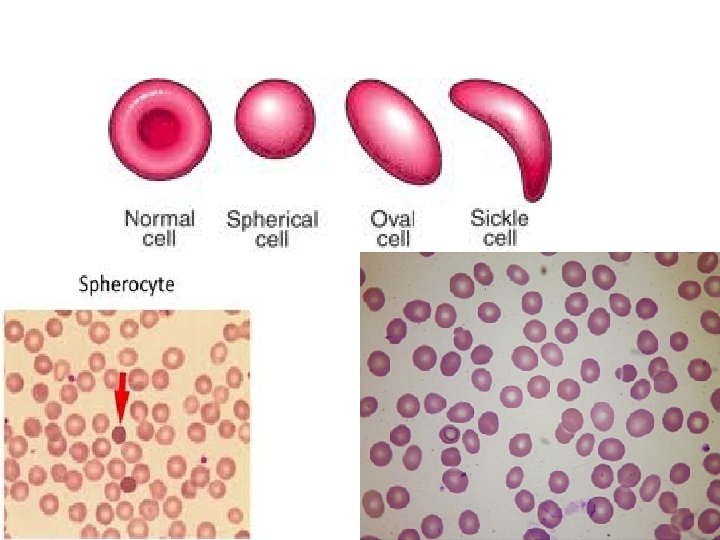

• When there is a family history it is usually easy to suspect the diagnosis • In most cases, the diagnosis can be made on the basis of red cell morphology and of a test for osmotic fragility, In some cases, a definitive diagnosis can be obtained only by molecular studies demonstrating a mutation in one of the genes underlying HS. • This is usually carried out in laboratories with special expertise in this area.

Treatment: Hereditary Spherocytosis • We don't have a causal treatment for HS; i. e. , no way has yet been found to correct the basic defect in the membrane-cytoskeleton structure. • However, it has been apparent for a long time that the spleen plays a special role in HS through a dual mechanism. • On one hand, like in many other HAs, the spleen itself is a major site of destruction; on the other hand, transit through the splenic circulation makes the defective red cells more spherocytic and therefore accelerates their demise, even though lysis may take place elsewhere. • For these reasons, splenectomy has long been regarded as a prime, almost obligatory therapeutic measure in HS.
 are as follows. (1) Avoid")
• • • Therefore, current guidelines (not evidence-based) are as follows. (1) Avoid splenectomy in mild cases. (2) Delay splenectomy until at least 4 years of age, after the risk of severe sepsis has peaked. (3) Antipneumococcal vaccination before splenectomy is imperative, whereas penicillin prophylaxis postsplenectomy is controversial. (4) There is no doubt that HS patients often may require cholecystectomy, in which case the practice has been to also carry out a splenectomy at the same time.
 • HE is at least as heterogeneous as HS, both from")
Hereditary Elliptocytosis (He) • HE is at least as heterogeneous as HS, both from the genetic point of view and from the clinical point of view. • Again it is the shape of the red cells that gives the name to these conditions, but there is no direct correlation between the elliptocytic morphology and clinical severity. • Clinical features and recommended management are similar to those outlined above for HS. • Although the spleen may not have the specific role it has in HS, in severe cases splenectomy may be beneficial.
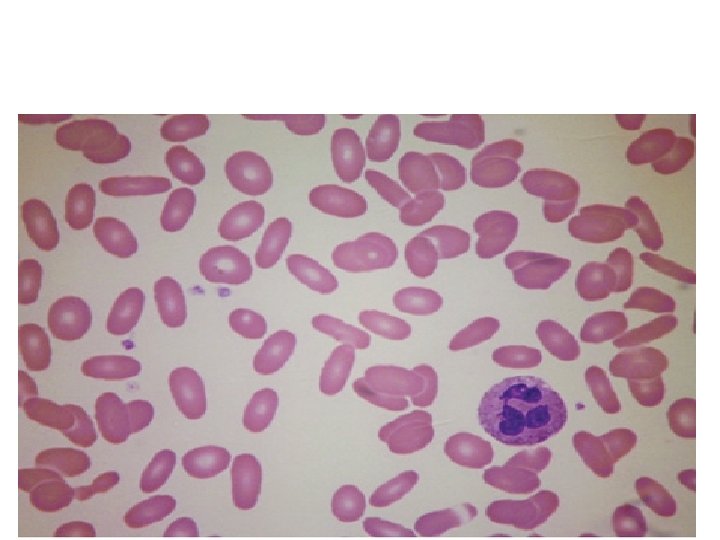

Hereditary Defects in Membrane Permeability • Hereditary Xerocytosis • The hallmark of this rare autosomal dominant disorder is an alteration in red cell membrane cation permeability that leads to a net loss of intracellular cations and water and to cell dehydration. • The molecular defect responsible for this phenotype remains to be elucidated, although the gene for a subset of xerocytosis has been mapped to the long arm of chromosome 16. • The dehydrated red cells appear on the blood smear as target cells or spiculated acanthocytes, and the increased MCHC leads to relatively non -deformable cells that can be sequestered and removed by the spleen. • The differential diagnosis of dehydrated red cells also includes the much more common sickle cell syndromes, hereditary spherocytosis, and hemoglobin C disease. • Splenectomy is relatively contraindicated in patients with hereditary xerocytosis because of the high risk of postprocedure thrombosis in these patients.

• Hereditary Stomatocytosis • This rare autosomal dominant disorder appears to be due to an inherited defect in Na+ permeability that leads to a net influx of Na+ and water and to cell swelling. • Several molecular defects are probably responsible for this phenotype because some families with this disorder experience severe hemolytic anemia whereas others have clinically mild disease. • The swollen red cells appear on the blood smear to have a mouth-like invagination in the membrane and are therefore called stomatocytes. • The differential diagnosis of stomatocytosis also includes the much more common acquired effects of acute alcoholism or liver disease. • Patients with the severe form of hereditary stomatocytosis often respond well to splenectomy, but some patients have developed hypercoagulability and catastrophic thrombosis after splenectomy.


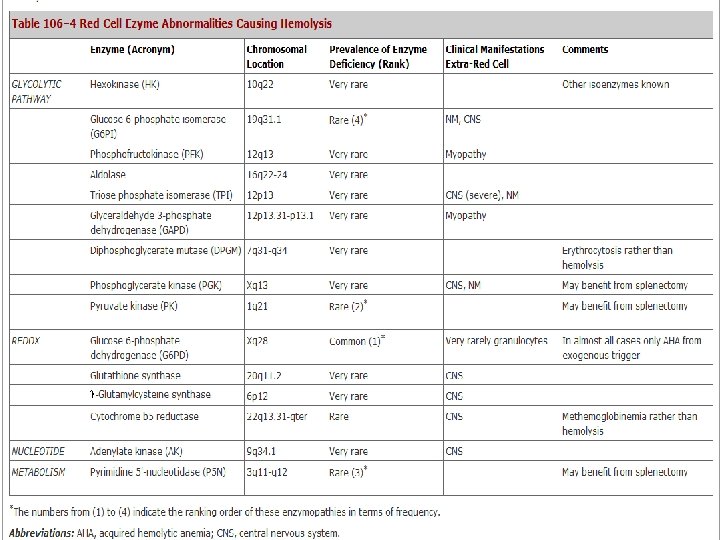
Abnormalities Pathway of the Glycolytic

• Since red cells, in the course of their differentiation, have sacrificed not only their nucleus and their ribsomes but also their mitochondria, they rely exclusively on the anaerobic portion of the glycolytic pathway for producing energy in the form of ATP. • Most of the ATP is required by the red cell for cation transport against a concentration gradient across the membrane. • If this fails, due to a defect of any of the enzymes of the glycolytic pathway, the result will be hemolytic disease

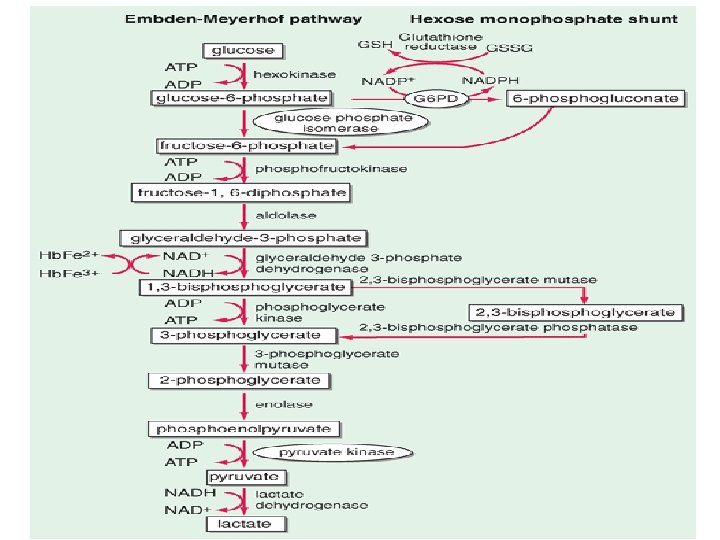
• Normal mature red cells have lost the cellular machinery responsible for oxidative phosphorylation, and the metabolism in these cells is almost entirely anaerobic. • The major red cell energy source is glucose, which is metabolized primarily by the glycolytic pathway (also called the Embden-Meyerhof pathway) and secondarily by the pentose phosphate pathway (also called the hexose monophosphate shunt)
 generates ATP for energy and membrane maintenance. •")
• The Embden-Meyerhof pathway (glycolysis) generates ATP for energy and membrane maintenance. • The generation of NADPH maintains hemoglobin in a reduced state. • The hexose monophosphate shunt generates NADPH that is used to reduce glutathione, which protects the red cell against oxidant stress. • Regulation of 2, 3 -bisphoglycerate levels is a critical determinant of oxygen affinity of hemoglobin.

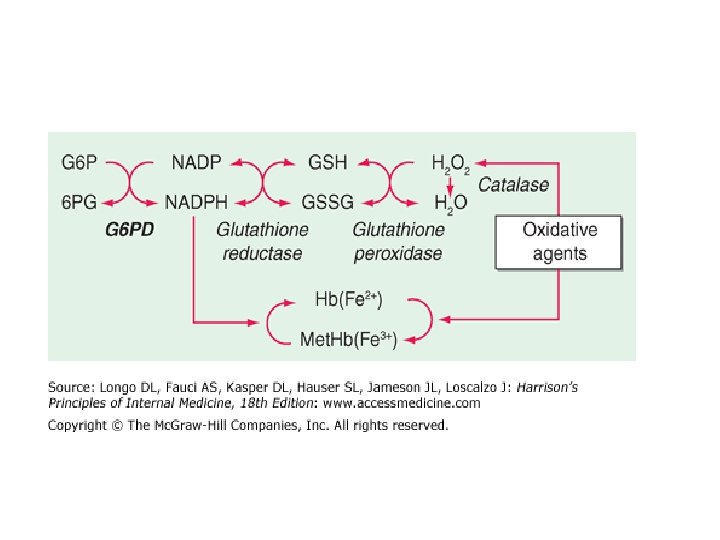
• NADPH is an essential cofactor for the enzyme glutathione reductase, which maintains glutathione in the reduced state necessary for detoxification of toxic oxygen products such as superoxide anion (O 2 -), hydrogen peroxide (H 2 O 2), and hydroxyl radical (. OH). • Normal red cells are continually subjected to these products as a result of intracellular heme oxidation. • In addition, certain drugs can markedly enhance oxidant generation by red cells, and many infections can induce oxidant generation by phagocytic cells in the circulation. • In the absence of reduced glutathione, toxic oxygen products can damage red cell lipids and proteins and result in hemolysis. Under conditions of oxidative stress

• Enzyme deficiency states in order of prevalence: glucose-6 -phosphate dehydrogenase (G 6 PD) > pyruvate kinase > glucose-6 -phosphate isomerase > rare deficiencies of other enzymes in the pathway. • The more common enzyme deficiencies are encircled in the following diagram.
 is a")
G 6 PD Deficiency • Glucose 6 -phosphate dehydrogenase (G 6 PD) is a housekeeping enzyme critical in the redox metabolism of all aerobic cells. • In red cells its role is even more critical, because it is the only source of NADPH that directly and via glutathione (GSH) defends these cells against oxidative stress. • G 6 PD deficiency is a prime example of an HA due to interaction between an intracorpuscular cause and an extracorpuscular cause, because in the majority of cases, hemolysis is triggered by an exogenous agent. • Although a decrease in G 6 PD activity is noted in most tissues of G 6 PD-deficient subjects, the decrease is less marked than in red cells, and it does not seem to have a clinical impact.

• Genetic Considerations • The G 6 PD gene is X-linked, and this has important implications. • First, as males have only one G 6 PD gene, they must be either normal or G 6 PD-deficient. • By contrast, females, having two G 6 PD genes, can be either normal or carrier.

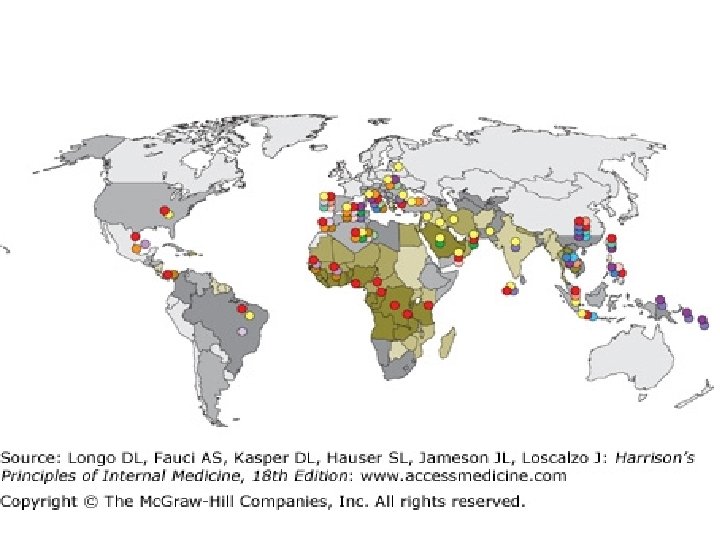
• G 6 PD deficiency is widely distributed in tropical and subtropical parts of the world (Africa, Southern Europe, the Middle East, Southeast Asia, and Oceania) and wherever people from those areas have migrated.

Clinical Manifestations • The vast majority of people with G 6 PD deficiency remain clinically asymptomatic throughout their lifetime; however, all of them have an increased risk of developing neonatal jaundice (NNJ), and a risk of developing acute hemolytic anemia (AHA) when challenged by a number of oxidative agents. • NNJ related to G 6 PD deficiency is very rarely present at birth. The peak incidence of clinical onset is between day 2 and day 3, and in most cases the anemia is not severe. • However, NNJ can be very severe in some G 6 PD-deficient babies, especially in association with prematurity, infection, and/or environmental factors (such as naphthalene-camphor balls used in babies' bedding and clothing), If inadequately managed, NNJ associated with G 6 PD deficiency can produce kernicterus and permanent neurologic damage.

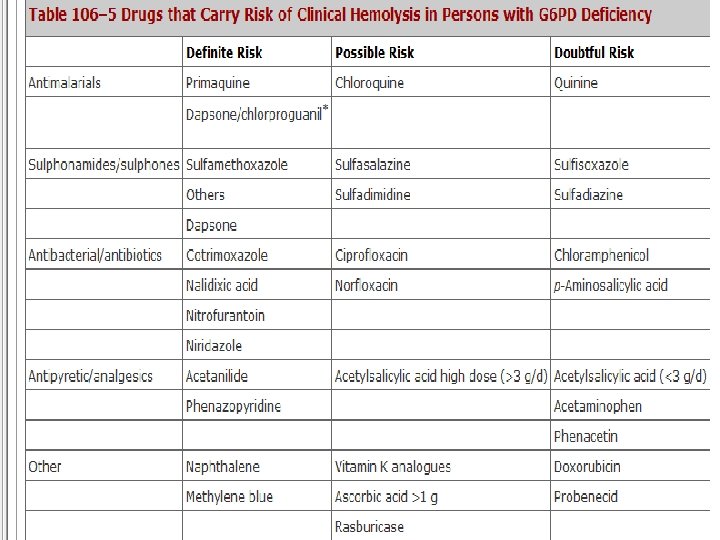
• • AHA can develop as a result of three types of triggers: (1) fava beans, (broad beans) (2) infections, and (3) drugs. • Typically, a hemolytic attack starts with malaise, weakness, and abdominal or lumbar pain. After an interval of several hours to 2– 3 days, the patient develops jaundice and often dark urine, due to hemoglobinuria. • • • The onset can be extremely abrupt, especially with favism in children. The anemia is from moderate to extremely severe. It is usually normocytic and normochromic, and it is due partly to intravascular hemolysis. Hence, it is associated with hemoglobinemia, hemoglobinuria, high LDH, and low or absent plasma haptoglobin(plasma glycoprotein binds to extracorpuscular Hb). And to a lesser extend extravascular


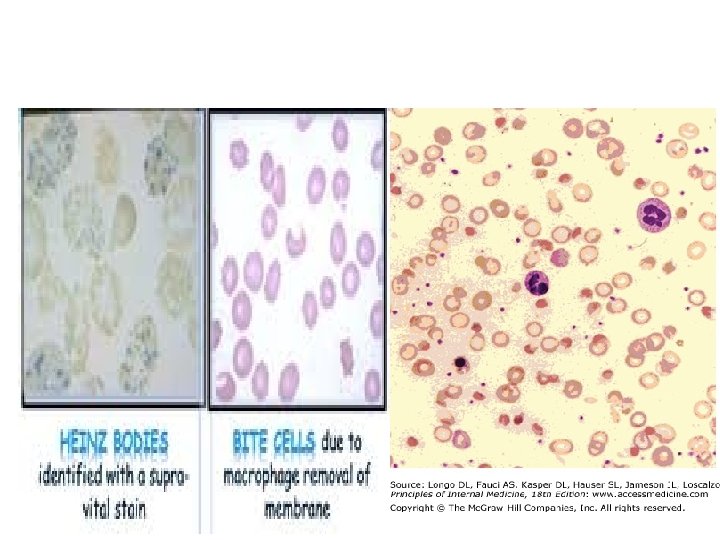
• The blood film shows anisocytosis, polychromasia. • The most typical feature is the presence of bizarre poikilocytes, with red cells that appear to have unevenly distributed hemoglobin ("hemighosts") and red cells that appear to have had parts of them bitten away ("bite cells" or "blister cells"). • presence of Heinz bodies, consisting of precipitates of denatured hemoglobin and regarded as a signature of oxidative damage to red cells • The most serious threat from AHA in adults is the development of acute renal failure (this is exceedingly rare in children). • Once threat of acute anemia is over, and in the absence of comorbidity, full recovery from AHA associated with G 6 PD deficiency is the rule.

• Laboratory Diagnosis • The suspicion of G 6 PD deficiency can be confirmed by semiquantitative methods often referred to as screening tests, which are suitable for population studies and can correctly classify male subjects, in the steady state, as G 6 PD-normal or G 6 PD- deficient. • However, in clinical practice, a diagnostic test is usually needed when the patient has had a hemolytic attack. • This implies that the oldest, most G 6 PD-deficient red cells have been selectively destroyed, and young red cells, having higher G 6 PD activity, are being released into the circulation. • Under these conditions, only a quantitative test can give a definitive result.

• Treatment: G 6 PD Deficiency • The acute hemolytic anemia of G 6 PD deficiency is largely preventable by avoiding exposure to triggering factors of previously screened subjects. • Of course, the practicability and cost-effectiveness of screening depends on the prevalence of G 6 PD deficiency in each community. • Favism is entirely preventable in G 6 PD-deficient subjects by not eating fava beans. • Drug-induced hemolysis can be prevented by testing for G 6 PD deficiency before prescribing; in most cases, one can use alternative drugs. • When AHA develops and once its cause is recognized, in most cases no specific treatment is needed. However, if the anemia is severe, it may be a medical emergency, especially in children, requiring immediate action, including blood transfusion. • If there is acute renal failure, hemodialysis may be necessary, but if there is no previous kidney disease, recovery is the rule.

• if the anemia is chronic and not severe, regular folic acid supplements and regular hematologic surveillance will suffice. • It will be important to avoid exposure to potentially hemolytic drugs, and blood transfusion may be indicated when exacerbations occur, mostly in concomitance with intercurrent infection. • In rare patients, regular blood transfusions may be required, in which case appropriate iron chelation should be instituted. • Unlike in hereditary spherocytosis, there is no evidence of selective red cell destruction in the spleen; however, in practice, splenectomy has proven beneficial in severe cases.

Pyruvate Kinase Deficiency • • • Abnormalities of the glycolytic pathway are all inherited and all rare. Among them, deficiency of pyruvate kinase (PK) is the less rare, with an estimated prevalence of the order of 1: 10, 000. The clinical picture is that of an HA that often presents in the newborn with neonatal jaundice; the jaundice persists, and it is usually associated with a very high reticulocytosis. The anemia is of variable severity; sometimes it is so severe as to require regular blood transfusion treatment; sometimes it is mild, bordering on a nearly compensated hemolytic disorder. As a result, the diagnosis may be delayed, and in some cases it is made in young adults; for instance, in a woman, during her first pregnancy, when the anemia may get worse. treatment: Pryuvate Kinase Deficiency The management of PK deficiency is mainly supportive. In view of the marked increase in red cell turnover, oral folic acid supplements should be given constantly. Blood transfusion should be used as necessary, and iron chelation may have to be added if the blood transfusion requirement is high enough to cause iron overload. In these patients, who have more severe disease, splenectomy may be beneficial. There is a single case report of curative treatment of PK deficiency by bone marrow transplantation from an HLA-identical PK-normal sibling. This seems a viable option for severe cases when a sibling donor is available.
- Slides: 45