Hemolytic anemia Sudi maiteh objectives Hemolytic anemia general

Hemolytic anemia � Sudi maiteh

objectives � Hemolytic anemia general principles � SICKLE CELL DISEASE � AUTOIMMUNE, COLD AGGLUTININ, AND DRUG- INDUCED HEMOLYTIC ANEMIA � HEREDITARY SPHEROCYTOSIS � PAROXYSMAL NOCTURNAL HEMOGLOBINURIA � GLUCOSE-6 -PHOSPHATE DEHYDROGENASE DEFICIENCY

General principles � Hemolytic anemias are caused by decreased RBC survival from increased destruction of the cells. � Chronic vs. acute � Extravascular vs. Intravascular � Clinical Presentation : - The usual symptoms of anemia (Fatigue and weakness occur with mild disease , Dyspnea and later confusion ) - Usually the onset is sudden. “simple blood loss excluded” - Associated with jaundice and dark urine - Fever, chills, chest pain, tachycardia, and backache may occur if the intravascular hemolysis is particularly rapid.
,")
� Diagnosis: � Normal MCV , The reticulocyte count is elevated (10– 20% range), � The LDH and indirect bilirubin are elevated. � peripheral smear abnormalities � haptoglobin maybe low with intravascular hemolysis � Hemoglobin and Hemosiderin , Hemosiderin may be present in the urine � There should not be bilirubin in the urine

�Treatment. Transfusion is needed as in all forms of anemia when the hematocrit becomes low. Hydration is, in general, useful to help prevent toxicity to the kidney tubule from the free hemoglobin

SICKLE CELL DISEASE �Pathology : Autosomal recessive hereditary disease �Hemoglobin S is due to a substitution of a valine for glutamic acid as the sixth amino acid of the beta globin chain. �Heterozygous form vs. Homorozygous form �A sickle cell acute painful crisis may be precipitated by hypoxia, dehydration, acidosis, infection, and fever. �Sickle cell crisis is usually not associated with an increase in hemolysis or drop in hematocrit.

Cont. � Clinical Presentation : � chronic : renal concentrating Defects, hematuria, bilirubin, Gallstones, osteomyelitis, retinopathy, recurrent infections from Pneumococcus or Haemophilus , splenomegaly (autosplenectomy) � Acute painful crisis : PAIN, � life-threatening manifestations of sickling : 1) acute chest syndrome : (severe chest pain, fever, leukocytosis, hypoxia, and infiltrates on the chest x-ray. ) 2) Stroke and TIA 3) Priapism 4) Blindness , myocardial infarction and cardiomyopathy 5) spontaneous abortion and low birth weight

Sickle trait gives normal hematologic picture with no anemia and a normal MCV. The only significant manifestation of trait is the renal concentrating defect presenting with isosthenuria and microscopic hematuria Increase risk for UTIs.

�Diagnosis: �mild to moderate anemia. �The hemoglobin electrophoresis is the most specific test. �The peripheral smear shows sickled cells. �The sickle prep �The white blood cell count is often elevated in the 10, 000– 20, 000 range.

� Treatment: � An acute sickle cell pain crisis is treated with fluids, analgesics, and oxygen. � Antibiotics(Ceftriaxone) are given with infection or even to patients with fever and leukocytosis. � In life threatening conditions : are managed with red blood cell transfusions if the hematocrit is low, and exchange transfusion if the hematocrit is high � Chronic management: � folic acid replacement and vaccinations � Hydroxyurea � Bone marrow transplantation

AUTOIMMUNE, COLD AGGLUTININ, AND DRUG-INDUCED HEMOLYTIC ANEMIA �can result from the production of Ig. G, Ig. M, or activation of complement C 3 against the red cell membrane �Cold ass. With Ig. M Vs. Warm ass. With Ig. G �Cause usually idopathic �Chronic deseases/drugs could induce it �The most common drugs are the penicillins, cephalosporins, sulfa drugs, quinidine, alphamethyldopa, procainamide, rifampin, and thiazides.

�Clinical Presentation : �The onset may be very sudden resulting in fever, syncope, congestive failure, and hemoglobinuria �Mild splenomegaly ( long duration ) �Cold agglutinin disease : cyanosis of the ears, nose, fingers, and toes. �Diagnosis : �The Coombs test is the specific test that diagnoses autoimmune �Spherocytes are often present on the smear

Cont, �Treatment: �Mild disease often occurs, which needs no treatment. In cases of drug-induced hemolysis, stop the offending drug �More severe autoimmune hemolysis is treated with steroids first. Splenectomy is done for those unresponsive to steroids. �Cold agglutinin disease is primarily managed by �avoiding the cold �Rituximab

HEREDITARY SPHEROCYTOSIS �Pathology : autosomal dominant disorder where the loss of spectrin in the red cell membrane �Clinical Presentation : �A chronic disorder with mild to moderate symptoms of anemia. �there is often splenomegaly and jaundice.
 is elevated. �a")
Cont, �Diagnosis: �osmotic fragility test. �The mean corpuscular hemoglobin concentration (MCHC) is elevated. �a negative Coombs test. �Treatment: �Most patients require no treatment beyond folate replacement chronically. �more severe anemia : spleen removal
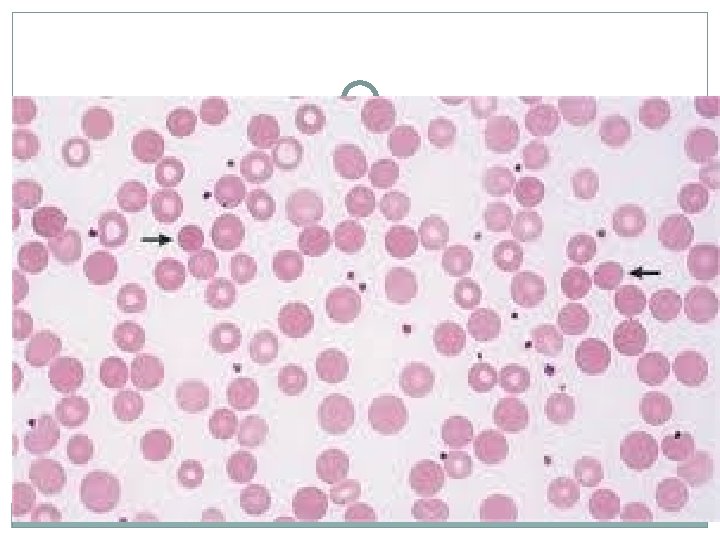

PAROXYSMAL NOCTURNAL HEMOGLOBINURIA �Pathology : A red cell membrane defect in phosphatidyl-inositol glycan A (PIGA) allows increased binding of complement to the red cell, leading to increased intravascular hemolysis. �Clinical Presentation: �Thrombosis of major venous structures, particularly the hepatic vein is a common cause of death in these patients �Diagnosis : The gold standard test is flow cytometry for CD 55 and CD 59 on white and red cells. In PNH, levels are low or absent.

Treatment. � Some patients with few or no symptoms require only folic acid and possible iron supplementation. � The disease may progress : � prednisone is often given to slow the rate of red blood cell destruction. � In the patient with acute thrombosis, thrombolytic therapy (streptokinase) � Antiplatelet agents and avoid medication that increase the risk for thrombosis (OCPs) � Allogeneic bone marrow transplantation has been the mainstay of curative therapy for PNH �Eculizumab treat the symptoms

GLUCOSE-6 -PHOSPHATE DEHYDROGENASE DEFICIENCY �Pathology : X-linked recessive , G 6 PD defect leads to increase RBCs susceptibility to oxidative stress. �The most common type of oxidant stress is actually from infections, not drugs. �The most commonly implicated drugs are sulfa drugs, primaquine, dapsone, quinidine, and nitrofurantoin. Clinical Presentation : �A sudden, severe, intravascular hemolysis, jaundice, dark urine, weakness, and tachycardia. �The main clue : history of recent drug ingestion

�Diagnosis: �Heinz bodies, Bite cells are seen on smear. �The definitive test is the G 6 PD level, which can be falsely normal immediately after an episode of hemolysis. Hence, the level is best tested about 1 week after the event.
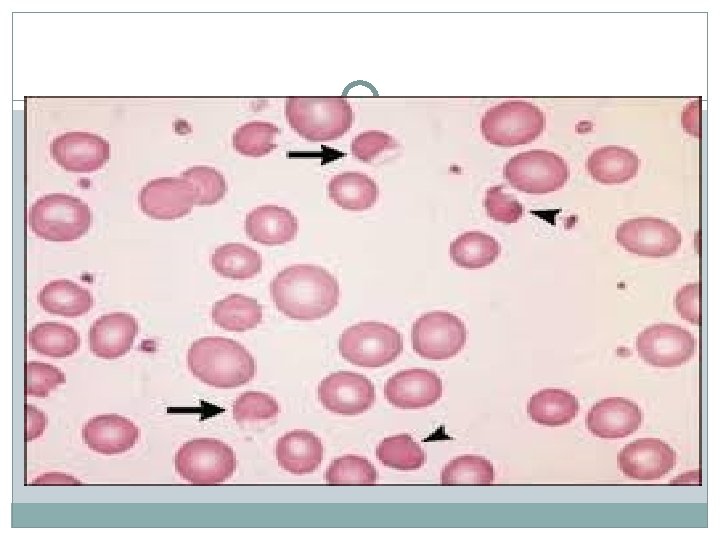

�Treatment: � There is no specific therapy beyond hydration and transfusion if the hemolysis is severe. �The main therapy is to avoid oxidant stress in the future.
- Slides: 22