HEMOGLOBINOPATHY Prof Dr Arzu SEVEN HEMOGLOBINOPATHY Mutations in

HEMOGLOBINOPATHY Prof. Dr. Arzu SEVEN

HEMOGLOBINOPATHY • Mutations in the genes that encode the α or β subunits of Hb potentially can affect its biological function • More than 800 known mutant human Hbs are both extremely rare and benign, with no clinical abnormalities • When a mutation compromises bilogic function hemoglobinopathy
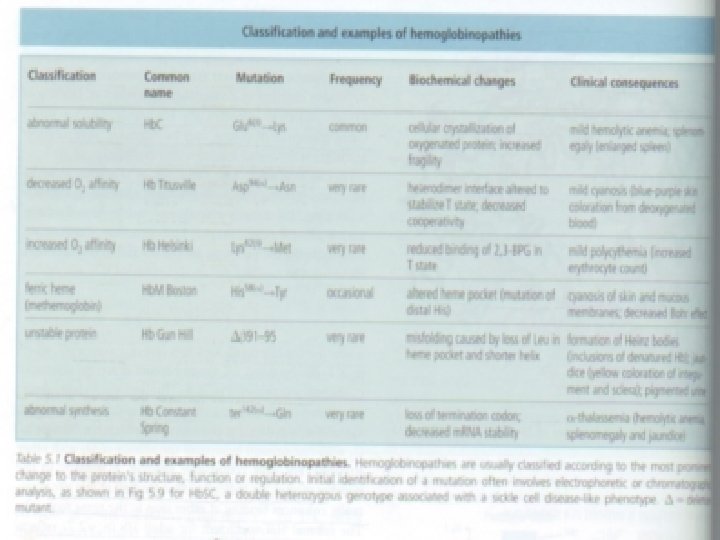

• Diagnosis of hemoglobinopathies • The mobility of a protein during elecrophoresis or chromatography is determined by its charge and interaction with matrix

• 3 commonly used techniques • Electrophoresis in agar gel at p. H: 6. 2 • IEF (using polyacrylamide gel) • Ion exchange chromatography

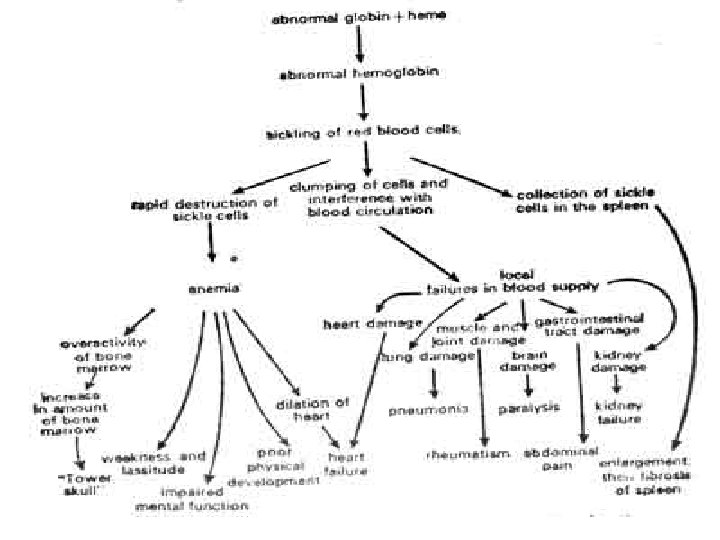
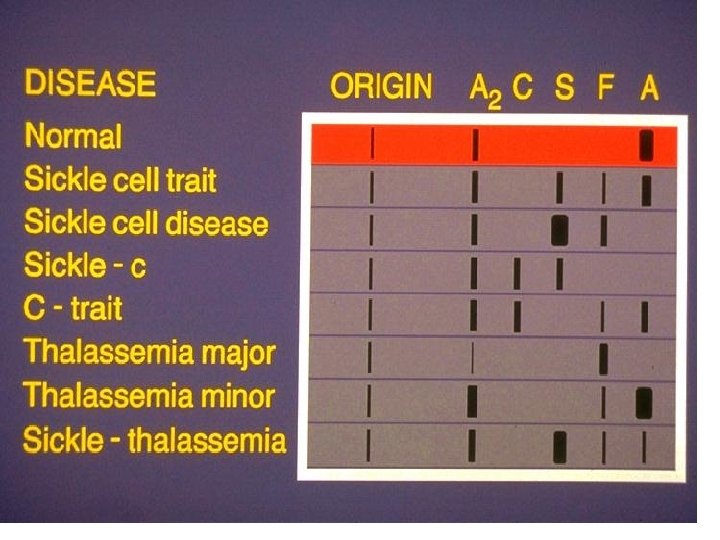
Sickling disorders=sickle cell disease Hb. S • Inherited, single point mutation in the gene encoding β_globulin • Glu Val • A surface-localized charged AA is replaced by a hydrophobic (nonpolar) residue • At low PO 2 deoxy Hb. S can polymerize to form long, insoluble fibers

• Sickle shape erythrocytes vulnerable to lysis • Hb. S, when deoxygenated, is less soluble it forms long, filamentous polymers that readily precipitate characteristic sickle shape
 the complex process of nucletion γ")
• In homozygous individual (Hb. S/Hb. S) the complex process of nucletion γ polymerization occurs readily • In heterozygous individual (Hb. A/Hb. S) sickle cell trait asymptomatic

• Sickled erythrocytes block blood flow especially in the spleeen γ joints cells lose water, become fragile, have shorter life span hemolysis γ anemia

• Intermittent episodes of hemolytic anemia • Acute vasoocclusive crises, impaired growth , increased susceptibility to infections, multiple organ damage • Heterozygosity is associated with an increased resistance to malaria, specifically growth of the infectious agent plasmodium falciparum in erythrocytes (selective advantage)
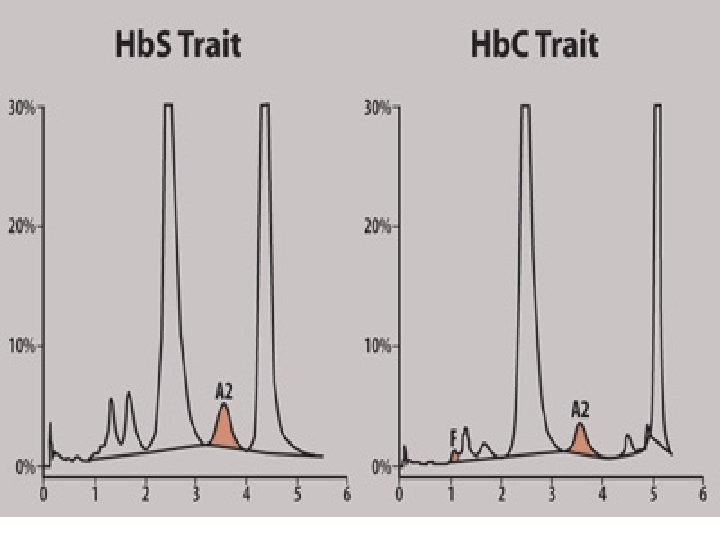
 • Copolymerize (interact) with Hb. S • when both are")
Hb. C (glu lys) • Copolymerize (interact) with Hb. S • when both are present, causing a sickling disorder resembling homozygous Hb. S disease • Hb. A , F and most Hb variants do not copolymerize with Hb. S they prevent severe sickling disorders when they are present with Hb. S

• When Hb. S trait is inherited together with β°_thalasemia trait severe sickling disorders • α_thalassemia are protective against severe sickling
 • • • Heme iron is ferric can neither bind")
Met Hbemia (Hb M) • • • Heme iron is ferric can neither bind nor transport O 2 Inherited due to met. Hb reductase deficiency (autosomal recessive) Acquired by ingestion of certain drugs (sulfonamides) γ chemicals
 Fe+3 makes a tight ionic complex with")
Hb. M: Histidin F 8 tyr (congenital) Fe+3 makes a tight ionic complex with phenolate anion of tyrosine • If α chain is affected: T state, O 2 affinity Bohr effect (-) • If β chain is affected: R_T switching Bohr effect(+)

• Infants are particulary vulnerable to met. Hbemi because Hb. F is more sensitive to oxidants compared to Hb A >%10 of Hb is in met. Hb cyanosis • Diagnosis: electrophoresis , characteristic absorption spectrum of met. Hb • Therapy: ingestion of methylene blue or ascorbic acid

Unstable Hb Hemolytic Anemia • More than 100 Hb variants show instability of either α or β globulin chain • Due to a substituon of a polar (or hydrophilic) AA for a nonpolar (or hydrophobic) AA that lines the pocket where heme is located • Köln Hbpati compensated hemolytic anemia • Zürich Hbpati sulfonamide_induced hemolysis

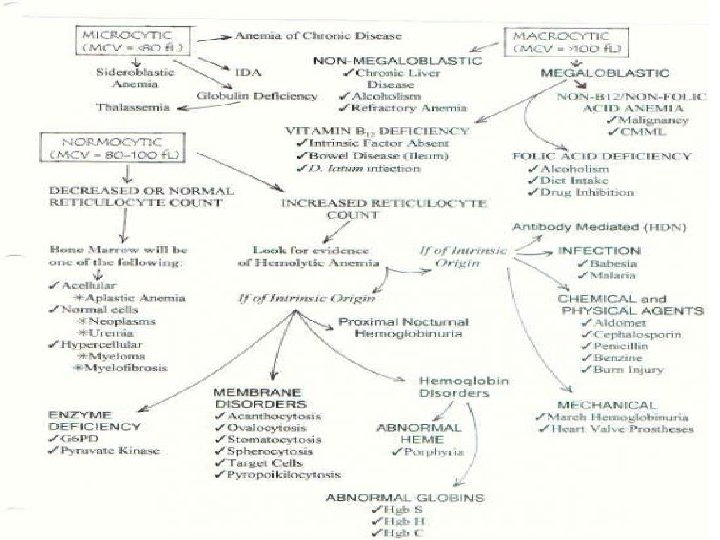
Thalassemias • Hereditory disorders characterized by a reduction in the synthesis of one type of globulin chain • α thalassemia: mutations in α-globulin genes(unequal crossingover γ large deletions) reduction in α chain synthesis

• β thalassemia: a very wide variety of mutations in β_globulin gene including deletions, nonsense γ frameshift mutations reduction in β chain synthesis

• • Thalasemia major: Severe anemia Hypochromic microcytic RBC Signs of accelerated hemolysis and regeneration (hyperbilirubiemia) • Hepato-splenomegali • Growth retardation • Bony abnormalities

• • • Thalassemia minor: Common γ mild condition Hypochromia Mild microcytosis of RBC Mild elevation of RBC Slight/no anemia

• Thalassemia trait: • Heterozygout state
- Slides: 25