HEMOGLOBINE Physiologie Hmolyse Globules rouges PRINCIPAUX CONSTITUANTS DU

HEMOGLOBINE Physiologie - Hémolyse

Globules rouges

PRINCIPAUX CONSTITUANTS DU GLOBULE ROUGE • Membrane érythrocytaire - Bicouche phospholipidique - Cholestérol - Protéines : glycophorines, spectrine, actine, ankyrine … - Importance du cytosquelette - groupes sanguins • Hémoglobine - Structure tétramérique representant 95% des protéines intracellulaires du GR - 4 chaînes de globines (protéines) (140 AA) 2 paires de chaînes polypeptidiques a, b ou d ou g - Groupe prosthétique de l’hème. Fer. Protoporphyrine Pigment coloré rouge

PRINCIPAUX CONSTITUANTS DU GLOBULE ROUGE • Equipement enzymatique Métabolisme énergétique - Absence de mitochondries - Voies glycolytique anaérobie Glucose … Pyruvate lactate ATP-dépendante, NAD-NADH Pyruvate kinase (PK) - Voie aérobie des pentoses phosphates Shunt NADPH dépendant Glucose phosphate déshydrogénase (G 6 PD)

Globules rouges Numération sanguine : 4 -6. 1012/L Réticulocytes : 20 -120. 1019/L Morphologie : petite cellule anucléée VGM: 90 fl Membrane = déformabilité -BBicouche lipidique -PProtéine 3, glycophorine -CCytosquelette : spectrine, actine Protéine 4. 1, ankyrine Hémoglobine Equipement enzymatique : -GGlycolyse : Voie principale anaérobie Pyruvate kinase Shunt des pentoses Glucose 6 phosphate deshydrogénase NADPH 2 -3 Diphosphoglycérate ATP = énergie Echanges gazeux

Structure de l’hémoglobine MM 64 k. Da 2 paires de chaînes polypeptidiques Contenant une porphyrine contenant un atome de FER

Structure quaternaire de l’hémoglobine adulte A a 2 b 2
 Protoporphyrine IX tétrapyrrolique FERREUX Histidine proximale Histidine distale O")
Structure de l’hème (hémoglobine, myoglobine) Protoporphyrine IX tétrapyrrolique FERREUX Histidine proximale Histidine distale O 2

Evolution de la synthèse des chaînes d’hémoglobine en fonction de l’âge

Hb. A 100 96 -98 % Hb. F <2% Hb. A 2 2 -3, 5 %

Différentes hémoglobines Période de la vie Hémoglobine A a 2 b 2 Vie Adulte A 2 a 2 d 2 Vie Adulte, (fœtale, néonatale) F a 2 g 2 Vie Fœtale, (fœtale, néonatale) Gower 1 z 2 e 2 Vie intrautérine précoce Gower 2 a 2 e 2 Vie intrautérine précoce Portland 1 z 2 g 2 Vie intrautérine précoce Portland 2 z 2 b 2 Vie intrautérine précoce

Gènes des globines Région régulatrice e g d b 5’ 3’ Chromosome 11 Région régulatrice z a 2 5’ a 1 3’ Chromosome 16 Sens de la transcription

Gènes de globine Bras p 5’ 3’ télomères Au cours du développement

Synthèse de la chaîne b Noyau Transcription Epissage ARNm Mitochondrie Ribosome Chaîne b Hème Chaîne a

ERYTHROPOIESE R-EPO R-Tr. F Hb CFU-GEMM IL-3, IL-4, IL 11 GM-CSF - + - BFU-E précoces IL-3, IL-9 GM-CSF +/- + +/- Colonies très volumineuses 14 -21 J Prolifération +++ BFU-E tardifs IL-3, IL-9 GM-CSF, EPO + + + Prolifération + Différenciation induction synthèse d’Hb CFU-E EPO +++ ++ + Colonies de moins de 50 cellules en 7 jours prolifération +/- Pro. Erythro blastes EPO +++ ++ ++ Subira 4 divisions pour donner naissance de 8 à 32 GR en 5/6 J Erythro basophile EPO ++ ++ ++ Apparition de l’hémoglobine Erythro polychrom +/- ++ ++ Erythro acidophile - ++ ++ Ne se divise plus Réticulocytes - + + Perte du noyau Coloration ARN GR - - + Perte de l’ARN

Propriétés chimiques ü Fixation, transport et délivrance de l’oxygène ü Egalement du CO 2 et ions H+ ü Forme T (tendu) de faible affinité et forme R (relâché) de forte affinité ü Passage de la forme T à la forme R par changement de conformation

Effet de l’oxygénation/déoxygénation sur la structure quaternaire de l’hémoglobine 2, 3 DPG b 2 a 2 b 1 a 1 x Structure déoxygénée Structure oxygénée

Courbes de dissociation de l’oxygène HEMOGLOBINE

Courbes de dissociation de l’oxygène Hb H, Hb Bart’s Hb A, A 2, F

Affinité de l’hémoglobine pour l’oxygène • Augmentation de l’affinité pour l’oxygène : – Hypothermie – Alcalose – Diminution du 2 -3 DPG • Diminution de l’affinité pour l’oxygène : – Fièvre – Acidose – Augmentation de 2 -3 DPG – Fixation du CO 2

Affinité de l’Hémoglobine F Hémoglobine A 2, 3 DPG b 2 a 2 Hémoglobine F b 1 g 2 O 2 a 1 a 2 a 1 x x Mère g 1 Foetus
 –")
Fonctions de l’hémoglobine • Transport de l’oxygène : – Hème : transport (Fe++) – Globine : hydrophobie, protection contre l’oxydation – Fe+++ : irréversible • Transport du CO 2 (10 %) – Carbamation groupes N-terminaux • Tampon • Transport d’oxide nitrique (NO) – Et effet « scavenger » : anti-oxydant N. B : Modification post-synthèse GLYCOSYLATION Hb. A 1 C

Hémolyse physiologique

Définition • Hémolyse = destruction physiologique des GR au terme de leur vie évaluée à 120 jours • 9 pour mille GR sont détruits par jour (= 50 ml de sang environ )

HEMOLYSE PHYSIOLOGIQUE • Durée de vie des GR : - 120 jours Mécanisme de la sénescence érythrocytaire - Déficit énergétique - Modification de la membrane Augmentation de la méthémoglobine et de l’affinité pour l’oxygène (déficit en 2 -3 DPG) • Modification des propriétés physiques, rhéologiques (déformabilité) du GR • Mécanismes de destruction des globules rouges sénescents: fragmentation de la membrane, lyse osmotique, phagocytose, cytolyse dépendant du complément
 Excrétion biliaire Métabolisme rénal et intestinal")
CATABOLISME DE L’HEMOGLOBINE Hème Bilirubine albumine Glycuro-conjugaison (foie) Excrétion biliaire Métabolisme rénal et intestinal (systèmes bactériens) Stercobilinogène (pigment brun) Urobilines
 - (Foie, rate: faible) - Système macrophagique de")
SITES DE L’HEMOLYSE • Extra-vasculaire (physiologique) - (Foie, rate: faible) - Système macrophagique de la moelle osseuse • Intra-vasculaire (pathologique) - Haptoglobine (a 2), synthèse hépatique - Complexes Hp-Hb (très forte affinité, irréversibles) - Clairance hépatique des complexes (MM 150 000) - Elimination urinaire de l’hémoglobine libre (hémoglobinurie). Réabsorbtion tubulaire saturable. Perte de fer

HEMOLYSE PATHOLOGIQUE Durée de vie courte Production médullaire Régénération Destruction Hémoglobine, Bilirubine Anémie « régénérative »
 • Anémies hémolytiques")
INTERET CLINIQUE • Comprendre la physiopathologie - des anémies hémolytiques (étiologies) • Anémies hémolytiques du nouveau-né • Transfusions érythrocytaires et hémovigilance

BIOLOGIE 1. Argument formel direct - Durée de vie des GR - Siège de destruction 2. Métabolisme de l’Hb - Haptoglobine - Bilirubine libre - Bilirubine glycuro-conjuguée - Hémoglobinurie - Hémoglobinémie 3. Métabolisme du fer - fer sérique - ferritinémie 4. Anémie, régénérative- réticulocytose 5. Morphologie érythrocytaire, VGM, IDR

Analyse de l’hémoglobine Électrophorèse de zone à p. H alcalin (p. H = 8, 6)

Analyse de l’hémoglobine Isoélectrofocalisation: fonction du p. Hi •
 Drépanocytose hétérozygote")
Analyse de l’hémoglobine Techniques électrophorétiques des chaînes d'hémoglobine en milieu dissociant (HPLC) Drépanocytose hétérozygote AS Hb. A 1 = 58 % Hb. S = 35, 3 % Hb. A 2 = 4, 7 % Non Hb = 0, 2 %

Analyse de l’hémoglobine Techniques de biologie moléculaire ü Laboratoires spécialisés ü Diagnostic prénatal ü Epidémiologie

MORPHOLOGIE DES GLOBULES ROUGES • • • Schizocytes Sphérocytes Dacryocytes Cellules-cibles Drépanocytes Destruction mécanique Anomalies de membrane « mécanique » , médullaire Hémoglobinopathies Hémoglobine S (gélification de l’Hb) • Stomatocytes • Elliptocytes • Acanthocytes Anomalies de membrane

Frottis normal

anisocytose

macrocytose
")
Microcytes (brulures graves)

Double population érythrocytaire

hypochromie

Hématies-cibles

polychromatophilie

Poïkilocytose

schizocytes

dacryocytes

Drépanocytose

ovalocytose

Corps de Jolly

Hématies ponctuées

Plasmodium

Agglutination des hématies

Hématies en rouleaux
 ü Anomalie de la chaîne de globine ü 1050 variants")
Anomalie qualitative (de structure) ü Anomalie de la chaîne de globine ü 1050 variants 260 pr le gène α 530 pr le gène β 70 pr le gène γ Ø Modification de la solubilité (Hb S, C, D, E) Ø Instabilité de la structure Ø Modification de son affinité pour l’O 2

Région régulatrice e g d b 5’ 3’ Chromosome 11 Région régulatrice z a 2 a 1 5’ 3’ Chromosome 16 Mutations : - faux sens (exon) : Hémoglobine S, C, E - introns/hors gène : certaines b thalassémies Délétions : - a thalassémies, certaines b/bd thalassémies

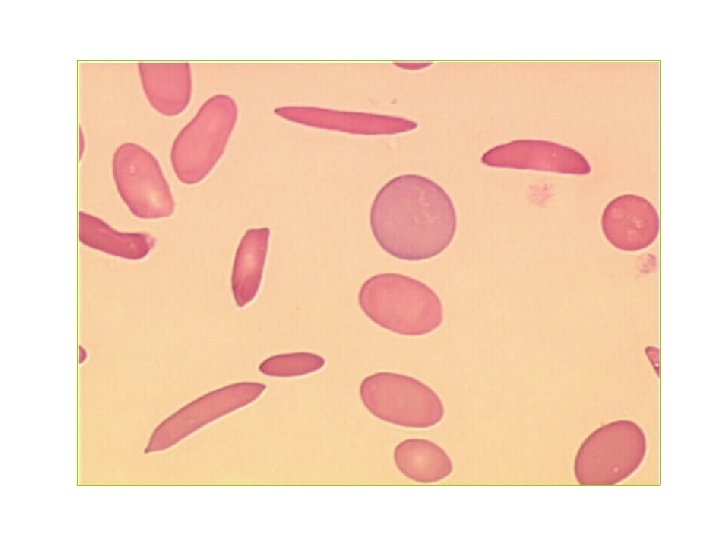
Drépanocytose Mutation faux sens : gène de la chaîne b globine Diminution de la Solubilité de la forme déoxygénée Polymérisation de l’hémoglobine FALCIFORMATION
 Contraste de Phase Polymérisation de l’Hb. S")
Drépanocytose Frottis sanguin (MGG) Contraste de Phase Polymérisation de l’Hb. S
 • Drépanocytose= état stable émaillé de complications aigues de fréquence et de")
Physiopathologie (drépanocytose) • Drépanocytose= état stable émaillé de complications aigues de fréquence et de sévérité variable d’un patient à l’autre • Cinétique polymérisation dépend de [Hb] intra-cellulaire, degré d’hypoxie, présence ou pas d’Hb F

Epidémiologie-génétique • Electivité ethnique +++ : – Populations noires d’Afrique (25 – 30 % porteurs du gène) – Antilles (10 %) – Bassin Méditerranéen – Proche Orient – Etats-Unis, puis Europe du Nord Transmission mode autosomique récessif Double hétérozygotie : SC, Sbthal, SE etc… fréquente
 • Anémie ± profonde 70 à 90 g/l")
Diagnostic biologique de la drépanocytose homozygote(1) • Anémie ± profonde 70 à 90 g/l • Normochrome, normocytaire • Frottis: anisocytose, poïkilocytose, corps de Jolly, drépanocytes, érythroblastose • Réticulocytose élevée • Signes biologiques d’hémolyse (frt, bilirubine, haptoglobine. . )
 • Test de falciformation positif • Test de solubilité (test d’Itano) faible")
Diagnostic biologique(2) • Test de falciformation positif • Test de solubilité (test d’Itano) faible solubilité Hb. S désoxy. • Diagnostic de certitude par 2 tests - isoélectrofocalisation et électrophorèse sur agar à p. H acide, ou HPLC pour quantification et identification Hb. A absente, Hb. S 75 à 95%, Hb. A 2 2 à 4%, HBF 1 à 15% • Biologie moléculaire
 : clinique • Avant 3 mois: Hb F • anémie hémolytique –")
Drépanocytose (S/S) : clinique • Avant 3 mois: Hb F • anémie hémolytique – ictère – splénomégalie ; atrophie splénique (asplénie) • crises douloureuses vaso-occlusives (fièvre, altitude, anesthésie, grossesse…) • sensibilité aux infections (pneumocoque) • manifestations viscérales – Diagnostic à l’occasion d’une complication aigue ou signe clinique particulier – Dépistage néonatal +++

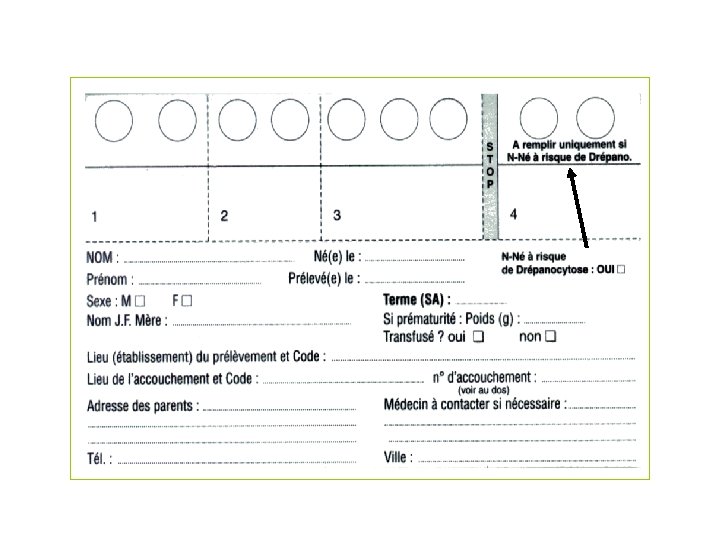
Dépistage néonatal en France Phénylcétonurie - Hypothyroïdie - Hyperplasie des surrénales - Drépanocytose - Mucoviscidose - • Intégré aux autres dépistages
 • SβThal: anémie, microcytose hypochromie,")
Diagnostic des doubles hétérozygotes • Autres syndromes drépanocytaires majeurs(SDM) • SβThal: anémie, microcytose hypochromie, Hb. S, Hb. A présente ou pas selon β° ou β+, Hb. F et Hb. A 2 • SC: cellules cibles, Hb. S=Hb. C, Hb. F

Drépanocytose hétérozygote • Non symptomatique • Mais possibilité de complications avec circonstances déclenchantes • Dépistage systématique familial • NFS normale, test de falciformation+ • Electrophorèse: Hb. A, Hb. S, Hb. A 2 • Conseil génétique
 Sud-Est asiatique (Kmers) hétérozygote assymptomatique")
Autres hémoglobinopathies • Hb E (béta 26 glu lys) Sud-Est asiatique (Kmers) hétérozygote assymptomatique homozygote : AH modérée svt bien tolérée (microcytose) • Hb C (béta 6 Glu Lys) Afrique haute Volta bien tolérée chez hétérozygote A/C • Hb D Punjab • Hb O Arab (balkans) • Variants asymptomatiques • Hémoglobines instables (présence de corps de Heinz)
 • (normale de la P 50=")
polyglobulies • Hémoglobines hyperaffines (haute affinité pour l’oxygène) • (normale de la P 50= 26 mm. Hg à 37°C) • DPG mutase • (normale du 2 -3 DPG= 15µM/g Hb) • Récepteur à l’EPO

Variations quantitatives Hb. A 2 ARNm d <<ARNm b • Diminution de l’Hb. A 2 : Carences en fer a/d et ad thalassémies • Augmentation de l’Hb. A 2 : b thalassémies Hb. F • Augmentation de l’Hb F : – Persistance héréditaire de l’Hb. F • Délétion des gènes d et b - b-thalassémie
")
Thalassémies: Epidémiologie • Défaut de synthèse d’une ou plusieurs chaines de globine (famille hétérogène) • αThalassémies : Sud-Est asiatique Afrique noire bassin méditerranéen Moyen Orient • βThalassémies : bassin méditerranée Sud-Est asiatique • Mais répartition plus grande du fait des migrations de populations

Diagnostic biologique βthal homozygote 1. Electrophorèse de l’Hb - Hb. F 50 à 95% - Hb. A 5 à 45% pour β+thal - Hb. A=0 pour β°thal - Hb. A 2 normale ou augmentée
 1. Mutation ponctuelle 2. sur 1 gène β =")
Génétique: transmission autosomique récessive(β thal) 1. Mutation ponctuelle 2. sur 1 gène β = βthal hétérozygote 3. sur 2 gènes β = βthal homozygote β°thal si pas de chaîne produite β+thal si diminution de synthèse 4. Rarement délétion β°thal 5. si délétion gène δ et γ = {β δ}thal 6. voir {β δ γ}thal si délétion étendue = persistance héréditaire Hb F(déficitβ compensé par γ) 7. Hb Lepore crossing over entre β et δ

α thalassémies • α thalassémie mineure • Perte 1 ou 2 gènes • Type 1 • Hb Bart’s Y 4 à la naissance 1à 2% ou 5% • Asymptomatique • Microcytose ou pseudo-polyglobulie • Adulte: parfois Hb. A 2<2, 5% • Diagnostic sur antécédents familiaux
 – Délétion 2 gènes: α°thalassémie (--/αα) hétérozygote")
Génétique: alpha thalassémie transmission autosomique récessive(α thal) – Délétion 2 gènes: α°thalassémie (--/αα) hétérozygote α+ thalassémie (-α/-α) homozygote – Délétion 1 gène: α+ thalassémie (-α/αα) hétérozygote – Délétion 3 gènes Hb. H β 4 Hémoglobinose H (--/-α) – Délétion 4 gènes: létale, anasarque Hb Bart’s tétramère γ 4

Hémolyses par anomalies membranaires du globule rouge • Sphérocytose héréditaire • Elliptocytose héréditaire (mutations chaînes de la spectrine ou protéine 4. 1) • Pyropoïkilocytose héréditaire (syndrome hémolytique grave en période néonatale) • Stomatocytose (anomalie d’un transporteur membranaire ionique) • Xérocytose héréditaire (anomalie contrôle des cations monovalents)

Hémolyses par déficits enzymatiques • Anomalies de la voie d’Embden. Meyerhof: • Pyruvate kinase • Glucose-phosphate isomérase • Autres déficits avec atteinte d’autres systèmes • Shunt des pentoses phosphates: G 6 PD • Voie de synthèse du glutathion

Déficit en Pyruvate Kinase • Epidémiologie: 1ère enzymopathie en Europe • Génétique: autosomique récessif • Physiopathologie: déficit de synthèse d’ATP inhibition de la pompe à sodium • Clinique : AH chronique (parfois ictère néonatal) splénomégalie • Rares complications: lithiase pigmentaire biliaire, hémochromatose • Biologie: dosage de PK

Déficit en G 6 PD • Epidémiologie: 5 à 25% Afrique subsaharienne, Asie tropicale, pourtour méditerranéen, 10% noirs américains • Génétique: récessif lié à l’X hommes 2 gènes normaux A et B (si mutés: A- chez noirs, méditerranéen dérive de B) • Biochimie: le G 6 PD muté est incapable de produire le NADPH nécessaire à la lutte contre oxydants dénaturation de l’Hb, précipitation (corps de Heinz) , lyse cellulaire

références • • Bonnes pratiques de l’étude de l’hémoglobine ABC 2003: 61; 401 -409 • Prise en charge de la drépanocytose chez l’enfant et l’adolescent (septembre 2005) http: //www. has-sante. fr • • • Référentiel « médicaments et déficit en G 6 PD » (25 février 2008) http: //www. agmed. sante. gouv. fr • • Syndromes thalassémiques majeurs et intermédiaires (juin 2008) http: //www. has-santé. fr
- Slides: 82