Hemoglobin metabolism diseases of hemoglobin T R form

Hemoglobin metabolism & diseases of hemoglobin
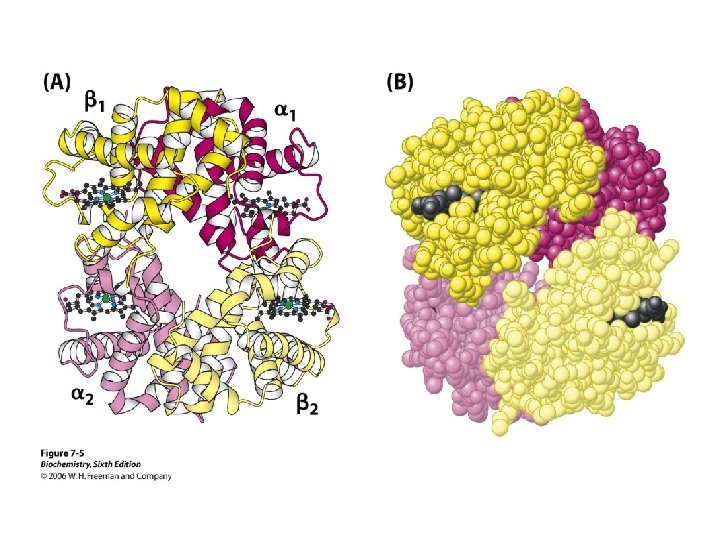


T & R form of Hb

Co-operative binding of oxygen

Binding of oxygen Mb Vs Hb

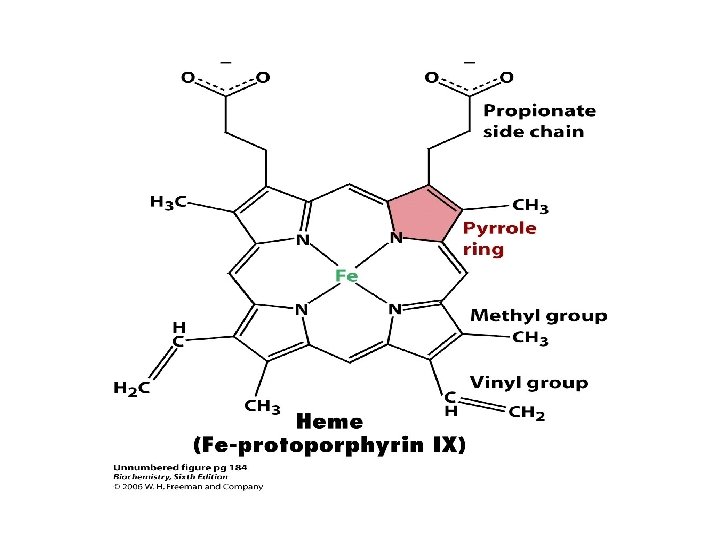
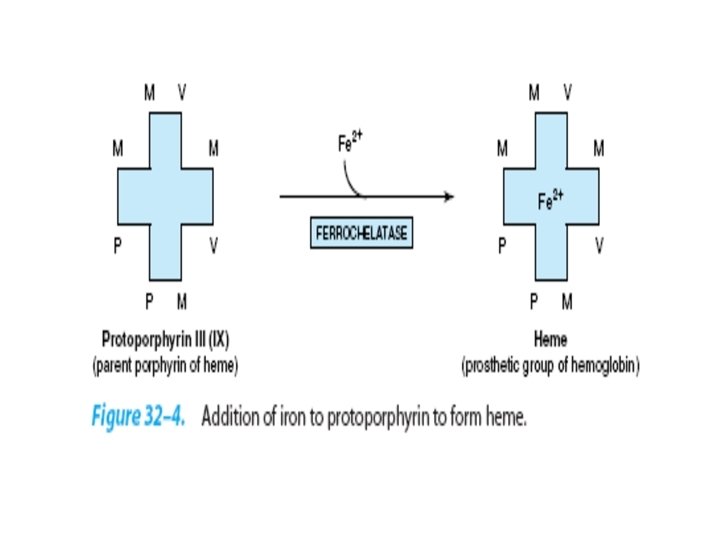


Transport of carbondioxide

Transport of carbondioxide

Bohr’s effect

Hemoglobinopathies • Hemoglobinopathies are the disorders caused by the synthesis of abnormal hemoglobin or insufficient production of normal hemoglobin or rarely both • Sickle cell hemoglobin: - abnormal hemoglobin • Thallasemias: - insufficient synthesis of hemoglobin

Sickle cell hemoglobin
 Mutation")
DEFINITION: When biological function is altered due to mutation in hemoglobin. CAUSES: (a) Mutation in structural gene Abnormalities in the primary sequence of globin chain. Exp. Hb-S, Hb-M, Hb-C, Hb-D, etc. (b) Mutation in regulatory gene Abnormalities in rate of synthesis. Exp. Thalassemias. 95% (single gene mutation/ point mutation) & others – frame shift mutation & terminator codon mutation EFFECT OF ABNORMAL HAEMOGLOBIN: • Abnormalities in red cell morphology. • Clinical manifestation haemolytic anaemia/ jaundice.

Genetic control of Hb synthesis 2 1 chrom 16 like gene G A chr-11 Normally synth of & ( in fetus) is carefully balance correct tetramer assembly
 Quantitative/ Structural gene defect E. g. Sickle cell anaemia (b) Qualitative/ Regulatory")
TYPES: (a) Quantitative/ Structural gene defect E. g. Sickle cell anaemia (b) Qualitative/ Regulatory gene defect E. g. Thalassaemias ( & ) STRUCTURAL HAEMOGLOBIN VARIANTS • Replacement/ alteration of single AA • Insertion/ deletion of AA or • Polypeptide fusion
 > 60 million carrier & 100, 000 affected infant")
SICKLE CELL ANAEMIA (Hb. S) > 60 million carrier & 100, 000 affected infant annually • Homozygous inheritance disorder PATHOPHYSIOLOGY: GENETIC DEFECT: Point/Frame shift GAG GUG [Glu acid valine] on 6 th position of chain of globin Hb. S
 Valine (hydrophobic/non-polar) • Less negative charge than")
BIOCHEMICAL EXPLANATION OF SICKLING • Glu (Hydrophilic/Polar) Valine (hydrophobic/non-polar) • Less negative charge than Hb. A (Hb surface charge). • localized stickiness on surface of chain. • deoxygenated Hb. S sticky patch bind with complementary patch polymerisation. • intracellular fibres are formed. • distortion of cells into sickle shaped. PREVENTION OF SICKLING: • Hb in oxygenated form or Deoxy Hb. TYPES: • (a) Homozygous – 80 -100% Hb. S & 0 -20% Hb. A. • (b) Heterozygous – may be asymptomatic 20 -40% Hb. S & 6080% Hb. A.

 • RBC more fragile (10 –")
SYMPTOMS: • Anaemia (6 – 8 g/d. L) • RBC more fragile (10 – 15 days) • Cells are rigid solubility obstruct the flow vasoocclusion tissue hypoxia. • Extreme pain & tissue death. • Susceptible to infection. RELATIONSHIP WITH OTHER DISEASES • Protection from malaria. • incidence to salmonella infection.

DIAGNOSIS • Sickling test • Electrophoresis
 Thalassemias - chain")
THALASSEMIAS Autosomal recessive disorders • Gene function is abnormal. TYPES: (a) Thalassemias - chain (b) Thalassemias - chain

THALASSEMIAS • synth/ total absence of globulin • involve the genes HBA 1& HBA 2 and forms 4 THALASSEMIA • synth / total absence of globin chain ( 4) or toxic aggregates
 DEFECTS: • Operator")
GENETICS: Defect in m. RNA for affected globin chain (quantitative/ qualitative) DEFECTS: • Operator gene/ Regulator gene defect. • Lowered stability of mutant m. RNA. • Loss of start signals for translation of m. RNA. • Non-sense mutations lead to premature chain termination. • Abnormal post transcriptional processing • Rapid degradation of highly unstable globin chain.
 Silent carrier 1")
Types of Thalassemia – Four types Type Missing gene Symptoms (a) Silent carrier 1 No (a) Thalassemia 2 Mild anaemia (a) Hemoglobin H Disease 3 Moderate anaemia (a) Hydrofetalis 4 Severe form
 Thalassemia MINOR ( Thalassemia Trait) – Heterozygous")
Types of Thalassemia – two types (a) Thalassemia MINOR ( Thalassemia Trait) – Heterozygous state – defect in only one globulin gene – Common in USA. – Usually asymptomatic. (b) Thalassemia MAJOR – Homozygous state (both gene) – At birth baby is healthy – After birth severe anemic & die 1 -2 yrs.

LABORATORY DIAGNOSIS OF HAEMOGLOBINOPATHIES • Hb%, Full blood count • Peripheral blood film • Electrophoresis • Peptide analysis • DNA finger printing technique
- Slides: 34