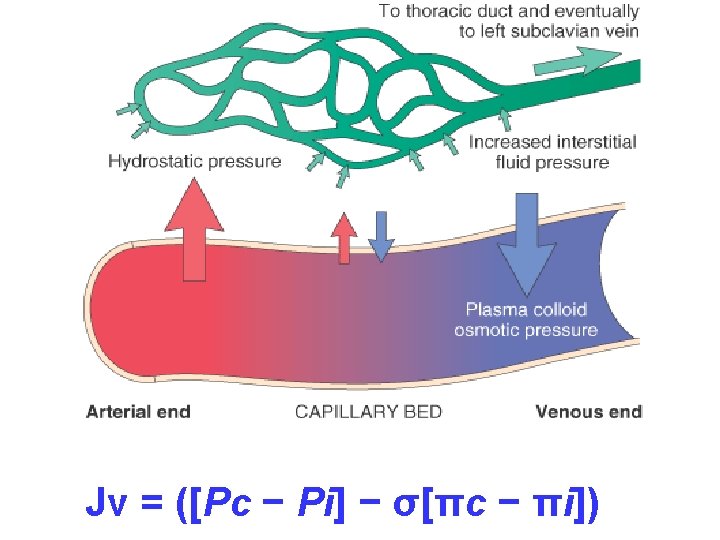
HEMODYNAMIC DISORDERS Jv Pc Pi c i Hemodynamic
![HEMODYNAMIC DISORDERS Jv = ([Pc − Pi] − σ[πc − πi])](https://slidetodoc.com/presentation_image_h/c5901c6c6a470b752aa6b3fbfabe15c1/image-1.jpg "HEMODYNAMIC DISORDERS Jv = ([Pc − Pi] − σ[πc − πi])")

 Hyperemia (INCREASED flow) Congestion")


 • • • Impaired venous return Congestive")
 • Protein-losing glomerulopathies (nephrotic syndrome) • Liver cirrhosis (ascites)")
 • Inflammatory • Neoplastic • Postsurgical • Postirradiation")

 • Chronic inflammation • Angiogenesis")



")
 “DEPENDENT” ANASARCA LEFT vs RIGHT HEART PERIORBITAL PULMONARY")

 – results from disturbance of Starling forces –")
")








 “DISSECTION”")

")


 ENDOTHELIUM • 2) PLATELETS • 3) COAGULATION “CASCADE”, or SEQ.")






")


 (prothrombotic) • THROMBIN")

/EXTRINSIC(Tiss. Fac) Proenzymes Enzymes Prothrombin(II) Thrombin(IIa) Fibrinogen(I) Fibrin(Ia) Cofactors")

PTT INTRINSIC (HEP Rx) PT (INR) EXTRINSIC (COUM Rx)")

 HYPERCOAGULATION, 1° 2 °")




 • COMMONEST: Factor V (Leiden) and Prothrombin defects •")
 Prolonged bed rest or immobilization Myocardial infarction")
 – ARTERY (OCCLUSIVE/INFARCT) – VEIN")

 • EMBOLIZATION • DISSOLUTION • ORGANIZATION • RECANALIZATION")

 V. T. • CHF a huge")



 • Fat • Air •")



 • AIR (SCUBA “bends”, sex? )")



")




 • HYPOVOLEMIC: (Hemorrhage or Leakage)")


")
 • • Peripheral vasodilation Pooling Endothelial Activation DIC *")


 • SYSTEMIC VASODILATION (hypotension) • ↓ MYOCARDIAL CONTRACTILITY •")
 • PROGRESSIVE (acidosis, early organ failure)")



 ACUTE TUBULAR NECROSIS (why?")





- Slides: 113
HEMODYNAMIC DISORDERS Jv = ([Pc − Pi] − σ[πc − πi])
• Hemodynamic Disorders • Thromboembolic Disease • Shock
Overview • • • Edema (increased fluid in the ECF) Hyperemia (INCREASED flow) Congestion (INCREASED backup) Hemorrhage (extravasation) Hemo-stasis* (opposite of thrombosis) Thrombosis (clotting blood) Embolism (downstream travel of a clot) Infarction (death of tissues w/o blood) Shock (circulatory failure/collapse)
EDEMA= ↑ECF • ONLY 4 POSSIBILITIES!!! –Increased Hydrostatic Pressure –Reduced Oncotic Pressure –Lymphatic Obstruction –Sodium/Water Retention
WATER • 60% of body • 2/3 of body water is INTRA-cellular • The rest is INTERSTITIAL • Only 5% is INTRA-vascular • EDEMA is SHIFT to the INTERSTITIAL SPACE FROM EITHER DIRECTION • CELL ECF VASC • HYDRO– -THORAX, -PERICARDIUM, -PERITONEAL • EFFUSIONS, ASCITES, ANASARCA
INCREASED HYDROSTATIC PRESSURE (i. e. , VENOUS) • • • Impaired venous return Congestive heart failure Constrictive pericarditis Ascites (liver cirrhosis) Liver is only a Portal-Caval filter? Venous obstruction or compression Thrombosis External pressure (e. g. , mass) Lower extremity inactivity with prolonged dependency Arteriolar dilation Heat Neurohumoral dysregulation
REDUCED PLASMA ONCOTIC PRESSURE (HYPOPROTEINEMIA) • Protein-losing glomerulopathies (nephrotic syndrome) • Liver cirrhosis (ascites) • Malnutrition • Protein-losing gastroenteropathy
LYMPHATIC OBSTRUCTION (LYMPHEDEMA) • Inflammatory • Neoplastic • Postsurgical • Postirradiation
Na+ RETENTION • Excessive salt intake with renal insufficiency • Increased tubular reabsorption of sodium • Renal hypoperfusion Increased renin-angiotensinaldosterone secretion
INFLAMMATION • Acute inflammation (r, c, d, T) • Chronic inflammation • Angiogenesis
CHF EDEMA-2 REASONS • INCREASED VENOUS PRESSURE DUE TO FAILURE • DECREASED RENAL PERFUSION, triggering of RENINANGIOTENSION-ALDOSTERONE complex, resulting ultimately in SODIUM RETENTION
HEPATIC ASCITES- 2 REASONS • PORTAL HYPERTENSION • HYPOALBUMINEMIA
ASCITES
RENAL EDEMA- 2 REASONS • SODIUM RETENTION • PROTEIN LOSING GLOMERULOPATHIES (NEPHROTIC SYNDROME)
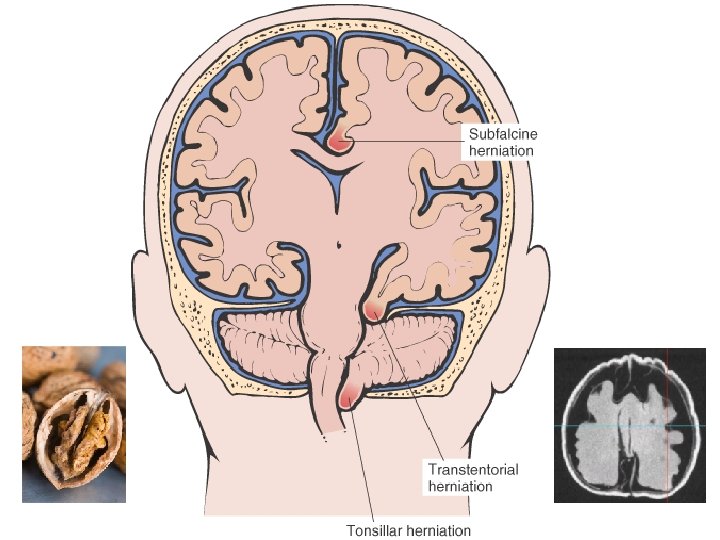
• • EDEMA SUBCUTANEOUS (“PITTING”) “DEPENDENT” ANASARCA LEFT vs RIGHT HEART PERIORBITAL PULMONARY CEREBRAL (closed cavity, no expansion) – HERNIATION of cerebellar tonsils – HERNIATION of hippocampal uncus over tentorium – HERNIATION, subfalcine
“Pitting” Edema
Transudate vs. Exudate • Transudate (water) – results from disturbance of Starling forces – specific gravity < 1. 012 – protein content < 3 g/dl, LDH LOW, ↓ Cells • Exudate (goo) – results from damage to the capillary wall – specific gravity > 1. 012 – protein content > 3 g/dl, LDH HIGH, ↑ Cells
HYPEREMIA/(CONGESTION)
HYPEREMIA Active Process CONGESTION Passive Process Acute or Chronic
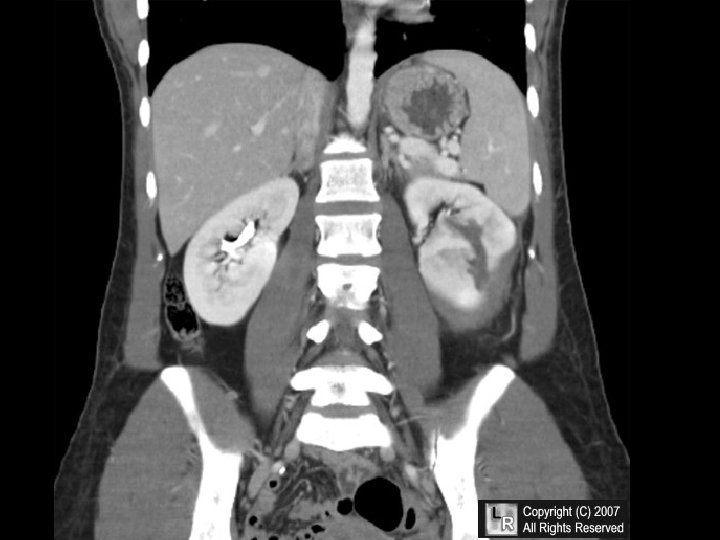
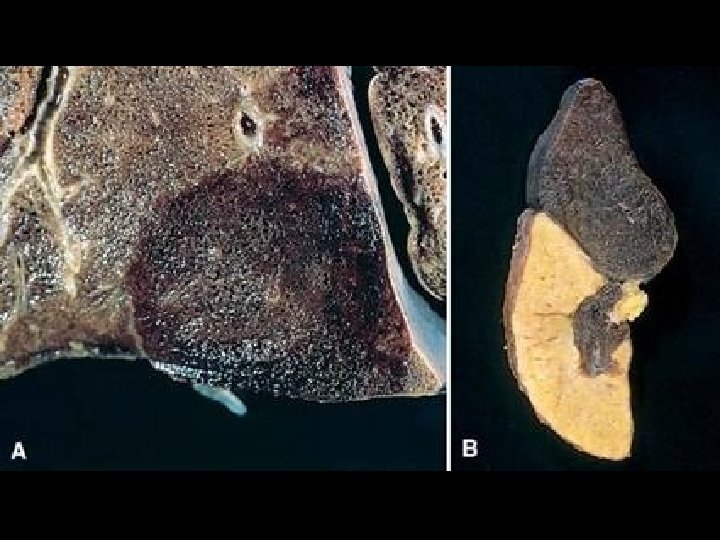
CONGESTION • LUNG, best example R. CHF – ACUTE – CHRONIC, hemosiderin • LIVER, best example L. CHF – ACUTE – CHRONIC, necrosis • CEREBRAL
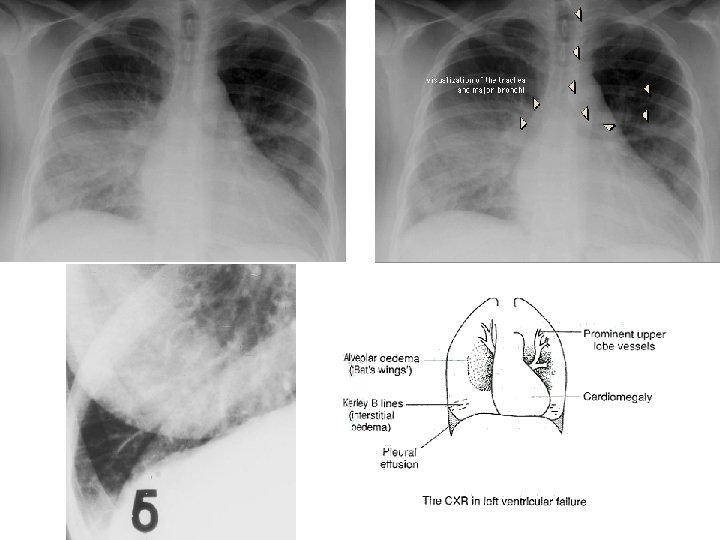
ACUTE PASSIVE HYPEREMIA/CONGESTION, LUNG septae alveoli
Kerley B Air Bronchogram
CHRONIC PASSIVE HYPEREMIA/CONGESTION, LUNG
Acute Passive Congestion, Liver
Acute Passive Congestion, Liver
CHRONIC PASSIVE HYPEREMIA/CONGESTION, LIVER
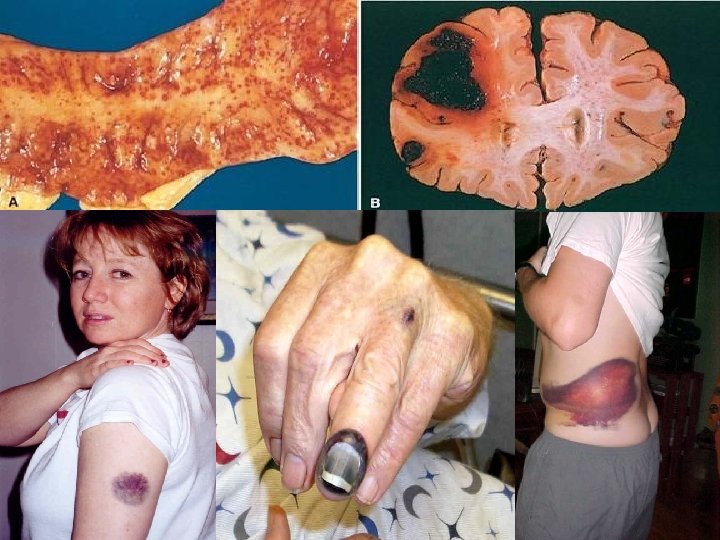
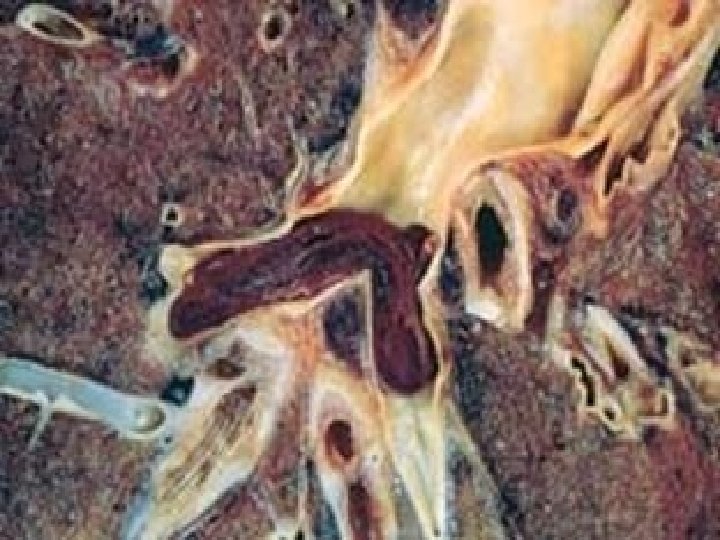
HEMORRHAGE • EXTRAVASATION beyond vessel • • “HEMORRHAGIC DIATHESIS” HEMATOMA (implies MASS effect) “DISSECTION” PETECHIAE (1 -2 mm) (PLATELETS) PURPURA <1 cm ECCHYMOSES >1 cm (BRUISE) HEMO-: -thorax, -pericardium, -peritoneum, HEMARTHROSIS • ACUTE, CHRONIC
EVOLUTION of HEMORRHAGE • ACUTE CHRONIC • PURPLE GREEN • HGB BILIRUBIN BROWN HEMOSIDERIN
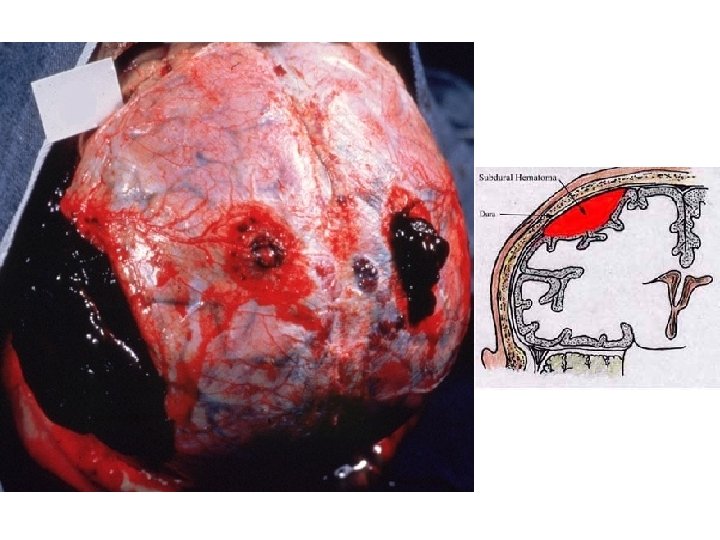
HEMATOMA vs. “CLOT” (Pre-mortem vs. Post-mortem)
HEMO”STASIS” • OPPOSITE of THROMBOSIS – PRESERVE LIQUIDITY OF BLOOD – “PLUG” sites of vascular injury • THREE COMPONENTS – 1 VASCULAR WALL, i. e. , endoth/ECM – 2 PLATELETS, “PRIMARY” COAG. – 3 COAGULATION CASCADE, or “SECONDARY” COAGULATION
SEQUENCE of EVENTS following VASCULAR INJURY • ARTERIOLAR VASOCONSTRICTION – Reflex Neurogenic – Endothelin, from endothelial cells • THROMBOGENIC ECM at injury site – Adhere and activate platelets – Platelet aggregation (1˚ HEMOSTASIS) • TISSUE FACTOR released by endothelium, plats. – Activates coagulation cascade thrombin fibrin (2˚ HEMOSTASIS) • FIBRIN polymerizes, TPA limits plug
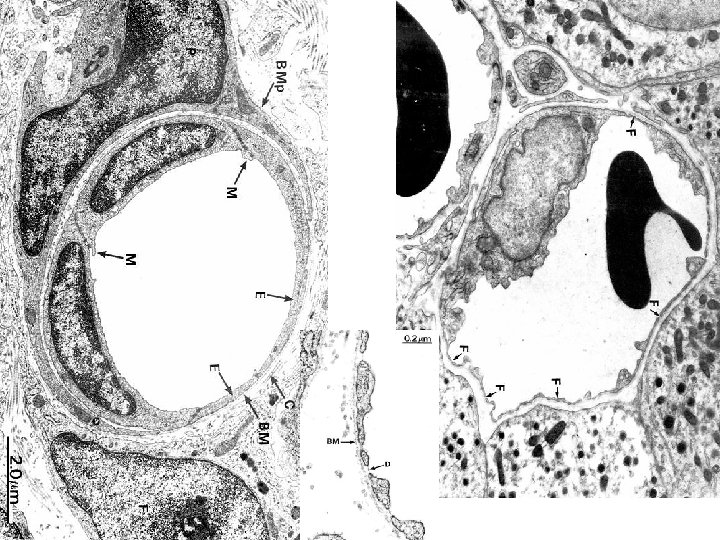
PLAYERS • 1) ENDOTHELIUM • 2) PLATELETS • 3) COAGULATION “CASCADE”, or SEQ.
ENDOTHELIUM • NORMALLY – ANTIPLATELET PROPERTIES – ANTICOAGULANT PROPERTIES – FIBRINOLYTIC PROPERTIES • IN INJURY – PRO-COAGULANT PROPERTIES
ENDOTHELIUM-JEKYLL • ANTI-Platelet PROPERTIES – Protection from the subendothelial ECM – Degrades ADP (inhib. Aggregation) • ANTI-Coagulant PROPERTIES – Membrane HEPARIN-like molecules – Makes THROMBOMODULIN Protein-C – TISSUE FACTOR PATHWAY INHIBITOR • FIBRINOLYTIC PROPERTIES (TPA)
ENDOTHELIUM-HYDE • PROTHROMBOTIC PROPERTIES – Makes v. WF, which binds Plats Coll – Makes TISSUE FACTOR (with plats) – Makes Plasminogen inhibitors
ENDOTHELIUM • • ACTIVATED by INFECTIOUS AGENTS ACTIVATED by HEMODYNAMICS ACTIVATED by PLASMA ACTIVATED by ANYTHING which disrupts it, including physical trauma “DISRUPTION”
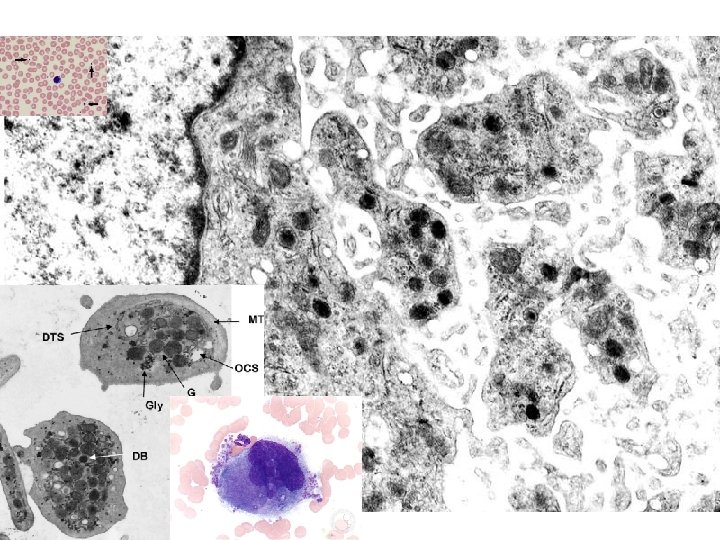
PLATELETS • ALPHA GRANULES – Fibrinogen – Fibronectin, a big CAM – Factor-V, Factor-VIII – Platelet factor 4 (anti-), TGF-beta • DELTA GRANULES (DENSE BODIES) – ADP/ATP, Ca+, Histamine, Serotonin, Epineph. • With endothelium, form TISSUE FACTOR
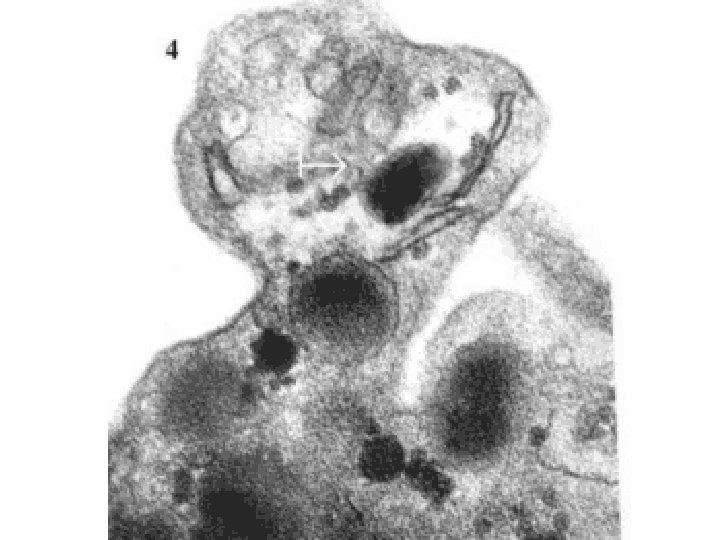
NORMAL platelet on LEFT, “DEGRANULATING” ALPHA GRANULE ON RIGHT AT OPEN WHITE ARROW
PLATELET PHASES • ADHESION • SECRETION (i. e. , “release” or “activation” or “degranulation”) • AGGREGATION
PLATELET ADHESION • Primarily to the subendothelial ECM • Regulated by v. WF, which bridges platelet surface receptors to ECM collagen
PLATELET SECRETION • BOTH granules, α and δ • Binding of agonists to platelet surface receptors AND intracellular protein “PHOSPHORYLATION”
PLATELET AGGREGATION • ADP • Tx. A 2 (Thromboxane A 2) (prothrombotic) • THROMBIN from coagulation cascade also • FIBRIN further strengthens and hardens and contracts the platelet plug
PLATELET EVENTS • • • ADHERENCE to ECM SECRETION of ADP and Tx. A 2 EXPOSE phospholip. Complexes Express TISSUE FACTOR PRIMARY SECONDARY PLUG STRENGTHENED by FIBRIN
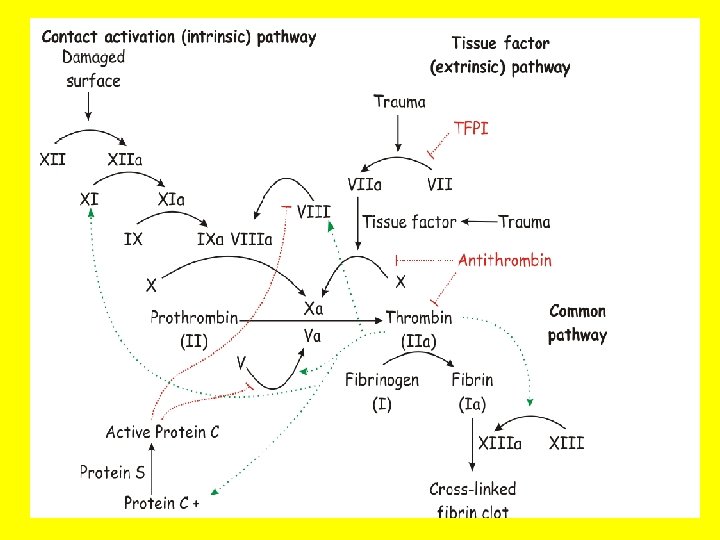
COAGULATION “CASCADE” • • • INTRINSIC(contact)/EXTRINSIC(Tiss. Fac) Proenzymes Enzymes Prothrombin(II) Thrombin(IIa) Fibrinogen(I) Fibrin(Ia) Cofactors – Ca++ – Phospholipid (from platelet membranes) – Vit-K dep. factors: II, VII, IX, X, Prot. S, C, Z
TF III
COAGULATION TESTS • • • (a)PTT INTRINSIC (HEP Rx) PT (INR) EXTRINSIC (COUM Rx) BLEEDING TIME (PLATS) (2 -9 min) Platelet count (150, 000 -400, 000/mm 3) Fibrinogen Factor assays
THROMBOSIS • Pathogenesis • • Endothelial Injury Alterations in Flow Hypercoagulability Morphology Fate Clinical Correlations Venous Arterial (Mural)
THROMBOSIS • Virchow’s TRIANGLE ENDOTHELIAL INJURY ABNORMAL FLOW (NON-LAMINAR) HYPERCOAGULATION, 1° 2 °
ENDOTHELIAL “INJURY” • Jekyll/Hyde disruption – any perturbation in the dynamic balance of the pro- and antithrombotic effects of endothelium, not only physical “damage”
ENDOTHELIUM • ANTI-Platelet PROPERTIES – Protection from the subendothelial ECM – Degrades ADP (inhib. Aggregation) • ANTI-Coagulant PROPERTIES – Membrane HEPARIN-like molecules – Makes THROMBOMODULIN Protein-C – TISSUE FACTOR PATHWAY INHIBITOR • FIBRINOLYTIC PROPERTIES (TPA)
ENDOTHELIUM • PROTHROMBOTIC PROPERTIES – Makes v. WF, which binds Plats Coll – Makes TISSUE FACTOR (with plats) – Makes Plasminogen inhibitors
ABNORMAL FLOW • NON-LAMINAR FLOW • TURBULENCE • EDDIES • STASIS • “DISRUPTED” ENDOTHELIUM ALL of these factors may bring platelets into contact with endothelium and/or ECF
˚ 1 HYPERCOAGULABILITY (INHERITED, GENETIC) • COMMONEST: Factor V (Leiden) and Prothrombin defects • Common: Mutation in prothrombin gene, Mutation in methylenetetrahydrofolate gene • Rare: Antithrombin III deficiency, Protein C deficiency, Protein S deficiency • Very rare: Fibrinolysis defects
˚ 2 HYPERCOAGULABILITY • • • (ACQUIRED) Prolonged bed rest or immobilization Myocardial infarction Atrial fibrillation Tissue damage (surgery, fracture, burns) Cancer (TROUSSEAU syndrome, i. e. , migratory thrombophlebitis) Prosthetic cardiac valves Disseminated intravascular coagulation Heparin-induced thrombocytopenia Antiphospholipid antibody syndrome (lupus “anti-”coagulant syndrome) • Lower risk for thrombosis: – – – Cardiomyopathy Nephrotic syndrome Hyperestrogenic states (pregnancy) Oral contraceptive use Sickle cell anemia Smoking, Obesity
MORPHOLOGY • ADHERENCE TO VESSEL WALL – HEART (MURAL) – ARTERY (OCCLUSIVE/INFARCT) – VEIN • OBSTRUCTIVE vs. NON-OBSTRUCTIVE • RED, YELLOW, GREY/WHITE • ACUTE, ORGANIZING, OLD
MURAL THROMBI, HEART
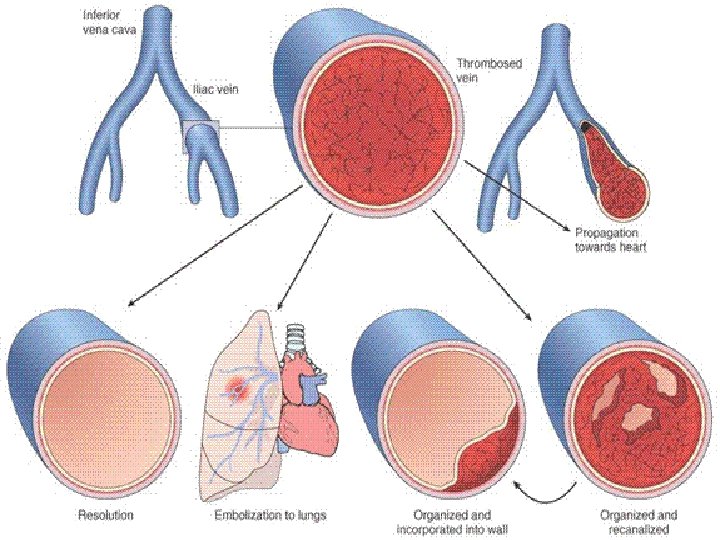
FATE of THROMBI • PROPAGATION (Downstream) • EMBOLIZATION • DISSOLUTION • ORGANIZATION • RECANALIZATION
OCCLUSIVE ARTERIAL THROMBUS
D. V. T. • D. (CALF, THIGH, PELVIC) V. T. • CHF a huge factor • INACTIVITY!!! (of lower extremities) • Trauma • Surgery • • Burns Injury to vessels, Procoagulant substances from tissues Reduced t-PA activity (Homeostasis shift)
ARTERIAL/CARDIAC THROMBI • ACUTE MYOCARDIAL INFARCTION = OLD ATHEROSCLEROSIS + FRESH THROMBOSIS • ARTERIAL THROMBI also may send fragments DOWNSTREAM, but these fragments may contain flecks of PLAQUE also • LODGING is PROPORTIONAL to the % of cardiac output the organ receives, i. e. , brain, kidneys, spleen, legs, or the diameter of the downstream vessel
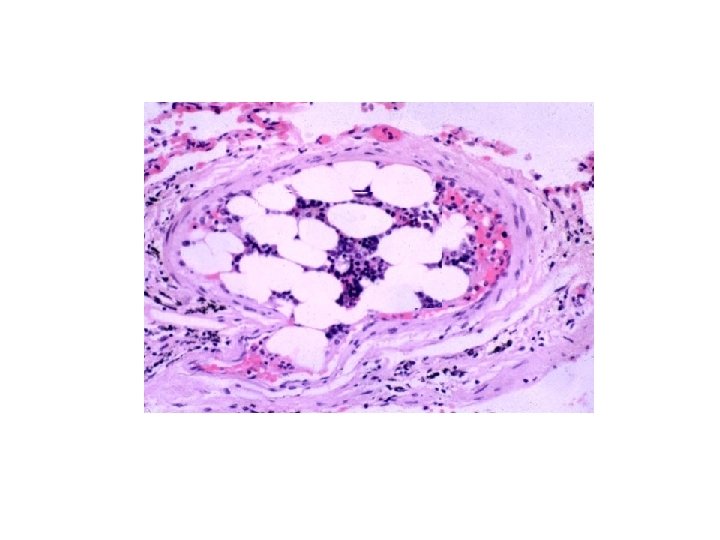
ATHEROEMBOLI • “CHOLESTEROL” clefts are components of atherosclerotic plaques, NOT thrombi!!!
Disseminated Intravascular Coagulation D. I. C. • OBSTETRIC COMPLICATIONS • ADVANCED MALIGNANCY • SHOCK (almost “synonymous” with DIC) NOT a primary disease CONSUMPTIVE coagulopathy, e. g. , reduced platelets, fibrinogen, F-VIII and other consumable clotting factors, brain, heart, lungs, kidneys, MICROSCOPIC ONLY
EMBOLISM • Pulmonary • Systemic (Mural Thrombi and Aneurysms) • Fat • Air • Amniotic Fluid
PULMONARY EMBOLISM • USUALLY SILENT • CHEST PAIN, LOW PO 2, S. O. B. • Sudden OCCLUSION of >60% of pulmonary vasculature, presents a HIGH risk for sudden death, i. e. , acute cor pulmonale, ACUTE right heart failure • “SADDLE” embolism often/usually fatal • PRE vs. POST mortem blood clot: – PRE: Friable, adherent, lines of ZAHN – POST: Current jelly or chicken fat: NON ADHERENT!!!
SYSTEMIC EMBOLI • “PARADOXICAL” EMBOLI due to R>L SHUNT • 80% cardiac/20% aortic • Embolization lodging site is proportional to the degree of flow (cardiac output) that area or organ gets, i. e. , brain, kidneys, legs
OTHER EMBOLI • FAT (long bone fx’s ) • AIR (SCUBA “bends”, sex? ) • AMNIOTIC FLUID, very prolonged or difficult delivery, high mortality
Amniotic Fluid Embolism
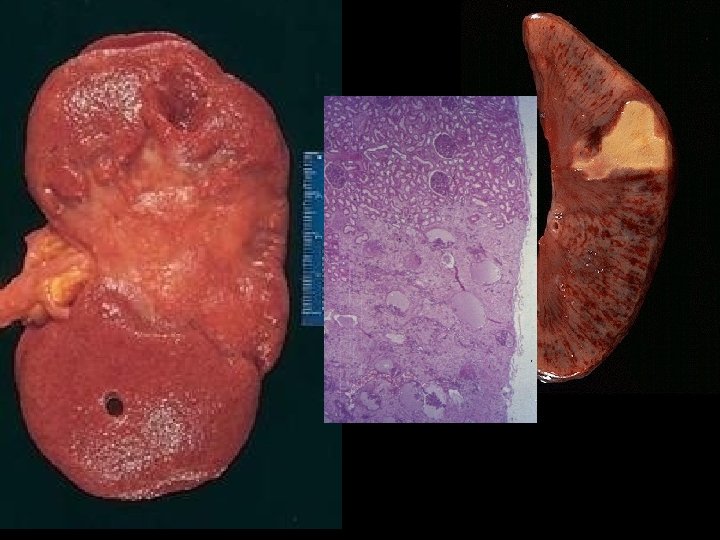
INFARCTION • Defined as an area of necrosis* secondary to decreased blood flow • HEMORRHAGIC vs. ANEMIC • RED vs. WHITE – END ARTERIES vs. NO END ARTERIES • ACUTE ORGANIZATION FIBROSIS
INFARCTION FACTORS • NATURE of VASCULAR SUPPLY • RATE of DEVELOPMENT – SLOW (BETTER) – FAST (WORSE) • VULNERABILITY to HYPOXIA – MYOCYTE vs. FIBROBLAST • CHF vs. NO CHF
HEART
SHOCK • Pathogenesis –Cardiac –Septic –Hypovolemic • Morphology • Clinical Course
SHOCK • Definition: CARDIOVASCULAR COLLAPSE • Common pathophysiologic features: – INADEQUATE CARDIAC OUTPUT and/or – INADEQUATE BLOOD VOLUME
GENERAL RESULTS • INADEQUATE TISSUE PERFUSION • CELLULAR HYPOXIA • UN-corrected, a FATAL outcome
TYPES of SHOCK • CARDIOGENIC: (Acute, Chronic Heart Failure) • HYPOVOLEMIC: (Hemorrhage or Leakage) • SEPTIC: (“ENDOTOXIC” shock, #1 killer in ICU) • NEUROGENIC: (loss of vascular tone) • ANAPHYLACTIC: (Ig. E mediated systemic vasodilation and increased vascular permeability)
CARDIOGENIC shock • • • MI VENTRICULAR RUPTURE ARRHYTHMIA CARDIAC TAMPONADE PULMONARY EMBOLISM (acute RIGHT heart failure or “cor pulmonale”)
HYPOVOLEMIC shock • • • HEMORRHAGE, Vasc. compartment H 2 O VOMITING, Vasc. compartment H 2 O DIARRHEA, Vasc. compartment H 2 O BURNS, Vasc. compartment H 2 O HEAT “STROKE”? (really loss of blood water due to severe dehydration)
SEPTIC shock • • • OVERWHELMING INFECTION “ENDOTOXINS”, i. e. , LPS (Usually Gm-) Gm+ FUNGAL “SUPERANTIGENS”, (Superantigens are polyclonal T-lymphocyte activators that induce systemic inflammatory cytokine cascades (storm? ) similar to those occurring downstream in septic shock, “toxic shock” antigents by staph are the prime example. )
SEPTIC shock events* (overwhelming infection) • • Peripheral vasodilation Pooling Endothelial Activation DIC * Think of this as a TOTAL BODY early inflammatory response
ENDOTOXINS • Usually Gm • Degraded bacterial cell wall products • Also called “LPS”, because they are Lipo- Poly-Saccharides • Attach to a cell surface antigen known as CD-14
ENDOTOXINS
SEPTIC shock events (linear sequence) • SYSTEMIC VASODILATION (hypotension) • ↓ MYOCARDIAL CONTRACTILITY • • • DIFFUSE ENDOTHELIAL ACTIVATION LEUKOCYTE ADHESION ALVEOLAR DAMAGE (ARDS) DIC VITAL ORGAN FAILURE CNS last
CLINICAL STAGES of shock • NON-PROGRESSIVE (compensatory mechanisms) • PROGRESSIVE (acidosis, early organ failure) • IRREVERSIBLE
NON-PROGRESSIVE • COMPENSATORY MECHANISMS • CATECHOLAMINES • VITAL ORGANS PERFUSED
PROGRESSIVE • HYPOPERFUSION • EARLY “VITAL” ORGAN FAILURE • OLIGURIA • ACIDOSIS
IRREVERSIBLE • HEMODYNAMIC CORRECTIONS of no use
PATHOLOGY • • MULTIPLE ORGAN FAILURE SUBENDOCARDIAL HEMORRHAGE (why? ) ACUTE TUBULAR NECROSIS (why? ) DAD (Diffuse Alveolar Damage, lung) (why? ) GI MUCOSAL HEMORRHAGES (why? ) LIVER NECROSIS (why? ) DIC (why? )
ARDS/DAD
MYOCARDIAL NECROSIS
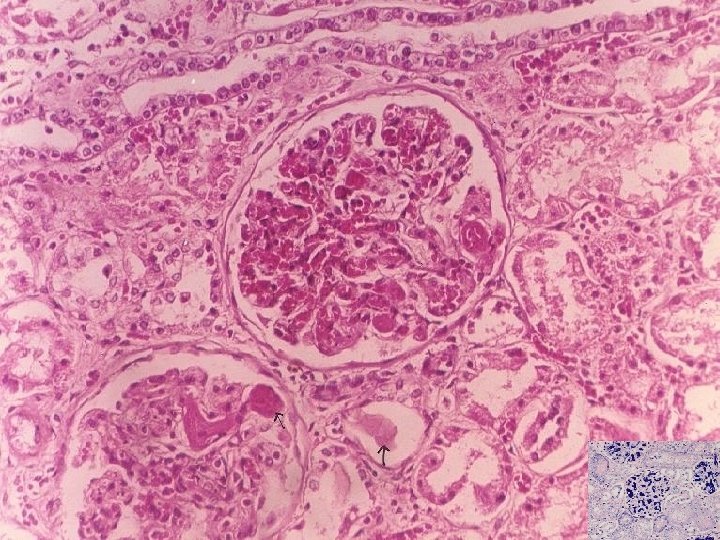
ATN
DIC
CLINICAL PROGRESSION of SYMPTOMS • • Hypotension Tachycardia Tachypnea Warm skin Cool skin Cyanosis Renal insufficiency Obtundance Death