Gentica Bioqumica Tipos de protenas Manuteno 90 presentes
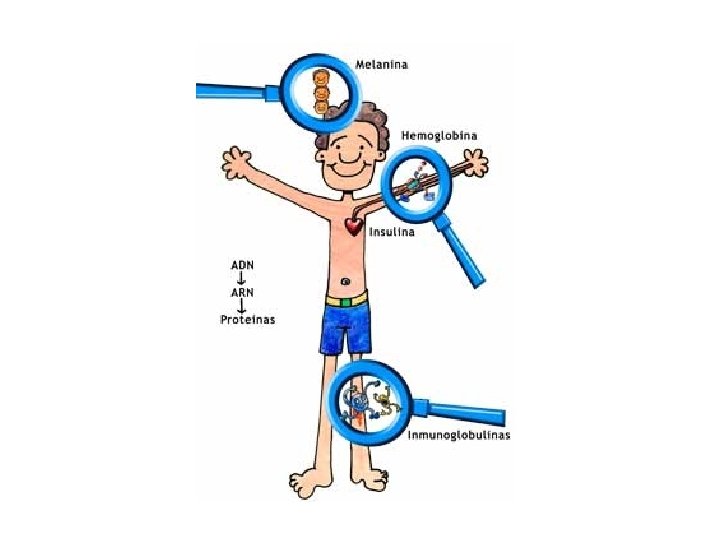

Genética Bioquímica

Tipos de proteínas • Manutenção – 90% – presentes em todas as células – estrutura e função da célula • Especialidade – 10% – produzidas por um número limitado de tipos celulares – funções singulares que contribuem para a individualidade da célula.

Classes funcionais de proteínas Enzimas Doença Herança Aminoácidos Fenilcetonúria AR Carboidratos Galactosemia AR Ácidos orgânicos Acidemia metilmalônica AR Lipídios Deficiência de carnitina AR Purinas Defic. de adenosinadesaminase AR Porfirinas Porfiria intermitente aguda AD

Transporte Entre órgãos Talassemia AR Membrana de organelas Cistinose AR Intracelular S. Menkes AR Membrana epitelial Fibrose cística AR

estrutural Extracelular Osteogênese imperfeita Membrana celular/ intracelular citoesqueleto S. Ehler-Danlos IV AD Organela S. Zellweger AR Hemostasia Hemofilia A XL Inibição de proteases Deficiência de alfa – 1 -antitripsina AR metabolismo Receptores de luz Retinite pigmentosa AD e Hormônios comunicação Defic. hormônio de crescimento AR Receptor de hormônio Leprechaunismo AR Receptor de metabólitos Hipercolesterolemia familiar AD homeostase extracelular intracelular AR, AD

P S C

Alteração estrutural das proteínas • As mutações dos genes estruturais reduzem a estabilidade da proteína. • A atividade de uma proteína depende de sua estrutura primária e eventos modificadores mediados por outras proteínas – modificações pós-tradução – síntese de polipeptídeos – moléculas que se associam à proteína para torná-la ativa como os co-fatores.

Anormalidades que afetam a estrutura da proteína • Primária – a conformação de uma proteína depende da estrutura primária de aminoácidos. – alterações da seqüência de aminoácidos alteram a conformação da proteína e reduzem sua estabilidade (anemia falciforme) • Secundária – durante ou após a tradução algumas proteínas sofrem modificações covalentes essenciais à sua função. – glicolisação (glicogenose) ou hidroxilação (S. Ehler-Danlos)

Anormalidades que afetam a localização/montagem da proteína • Depois que o peptídeo é dobrado em sua conformação correta, ele deve ser dirigido à sua posição celular ou extra-celular antes de assumir sua função. • Defeitos primários que comprometem o trânsito celular – acidúria metilmalônica - códon finalizador cria uma proteína que não é captada pela mitocôndria, impedindo a formação de um produto gênico funcionante. • Defeitos secundários que comprometem o trânsito celular – algumas proteínas são localizadas com base em sua estrutura conformacional pós-tradução. – As hidrolases ácidas na doença das células I apresentam um defeito pós-tradução que é essencial à sua ligação aos lisossomos.

Mutações que alteram a associação de proteínas • Mutações primárias que comprometem a montagem ou interação das subunidades em proteínas multiméricas – se uma proteína for composta de subunidades, uma mutação pode impedir a associação delas (osteogênese imperfeita tipo II)

• Deficiências secundárias devido à ausência de formação de complexos de múltiplas proteínas – a função de uma proteína pode sofrer redução secundária – peptídeos que formam um complexo tem defeitos genéticos primários que impedem sua síntese – a estrutura sofre proteólise prematura – os peptídeos funcionam mas numa posição errada onde sua função é neutra. – na doença de Zellweger há uma alteração na biogênese dos peroxissomos. As enzimas deficientes são sintetizadas normalmente mas sofrem degradação prematura no citosol, onde são vulneráveis à proteólise.

Mutações que comprometem a ligação, disponibilidade ou remoção de co-fatores • Algumas proteínas são ativadas após a sua ligação com cofatores não protéicos. • Mutações primárias podem comprometer a ligação do cofator à proteína (homocistinúria). • Anormalidades secundárias da função da proteína devida a inadequação da síntese, transporte, fixação ou remoção de moléculas (homocistinúria, deficiência de holocarboxilase-sintetase, biotinidase). • A natureza desses defeitos os torna possíveis de tratamento com a terapia bioquímica específica, principalmente quando o co-fator é uma vitamina hidrossolúvel.

Localização da proteína X sítio da fisiopatologia • A mutação de uma proteína específica de um tecido pode produzir uma doença restrita àquele tecido (talassemia) ou ter efeitos secundários sobre outros tecidos (fenilcetonúria) • Os efeitos clínicos das mutações de uma proteína de manutenção se limitam aos tecidos em que a proteína é mais abundantes e exerce uma função especializada. • No defeito do ciclo da uréia as enzimas argininosuccinato-sintetase e argininosiuccinato-liase estão em todas as células, mas seu nível de expressão é maior no fígado, local onde ocorre a conversão de amônia em uréia (função do ciclo da uréia). • Em algumas doenças a perda da função de manutenção é responsável por uma apresentação clínica e a perda da função específica do tecido produz outras manifestações – a deficiência grave de HPPT, causa S. de Lesh-Nyhan e moderada, gota e nefrolitíase sem alterações do sistema nervoso.

Anomalia molecular X Fenótipo • Mutações diferentes de um único gene podem produzir fenótipos clínicos diferentes. • A heterogeneidade de um lócus freqüentemente é responsável pela heterogeneidade clínica. • As apresentações clínicas de várias mutações podem ser tão diferentes que, apenas do ponto de vista clínico, não é evidente que afetem a mesma proteína. • Para algumas proteínas não é importante se a mutação reduziu a quantidade da proteína normal ou se resulta em quantidade normal de proteína anormal como na doença de Tay-Sachs. • Para outras essa alteração é importante como as que acarretam subprodução das cadeias de globina (talassemia) ou as mutações puntiformes que não reduzem a quantidade da cadeia de globina (metemoglobinemia).

acúmulo de moléculas complexas metabolismo energético sintomas permanentes/progres sivos sem relação com a dieta peroxissomos Produção/utilização deficiente de energia glicogenoses acidemias láticas doenças mitocondriais lisossomos intoxicação períodos assintomáticos intercalados com crises agudas aminoacidopatias acidúria orgânica defeito do ciclo da uréia

Suspeita clínica • consanguinidade • irmão com mesmo quadro clínico • ausência de afecção pediátrica justificável • acometimento súbito sem causa pediátrica aparente

Erros inatos do metabolismo • • CARBOIDRATOS LIPÍDIOS AMINOÁCIDOS ACIDEMIAS ORG NICAS • • CICLO DA URÉIA DDL METAL PURINAS
 • asfixia • doença respiratória do RN")
Diagnóstico diferencial • sepse (não exclui EIM) • asfixia • doença respiratória do RN

Achados predominantes • • • Encefalopatia aguda Doença cardíaca Doença hepática aguda DHE e DAB Dismorfismos

Doença de Tay-Sachs • Início dos sintomas – 6 meses, estado vegetativo com 1 -2 anos, óbito com 2 -3 anos • Incidência – Asquenazim (EUA) 1: 3. 900 e não asquenazim (EUA) 1: 112. 000 • Defeito básico – ausência de hex. A • Diagn. Prénatal – ensaio enzimático e análise DNA (chr. 15)

Fibrose Cística chr. 7 q 31 -32 250 Kb 27 exons CFTR (cystic fibrosis transmembrane condutance regulator) 170 Kd

Fibrose cística • • • Doença letal mais comum na população branca AR Defeito primário - regulação transporte de cloro Diagnóstico: IRT, Na/Cl no suor, testes biomol. QC: SP (15%): pneumonia de repetição (S. aureus, P. aeruginosa) - BCE, bronquiectasia IP (85%): desnutrição, infertilidade, cirrose hepática, íleo meconial (10 -20%), dilatação ductos gl. salivares (atrofia e fibrose)


Doença de Niemann-Pick • • Início da manifestação: 6 m • Hepatoesplenomegalia, deficit de crescimento e desenvolvimento, xantomas, febre, linfadenopatia, vômito, mácula cor de cereja • Tratamento – def. Transporte e esterificação de • colesterol intracelular Óbito: 1 -2 a atividade esfingomielinase – biópsia hepática e MO • Diagn. prénatal: análise de DNA • Defeito básico – def. Esfingomielinase- acúmulo esfingomielina e colesterol • Tipo a-b – def. Esfingomielinase (80%) • Tipo c-d

Gaucher Doença de depósito lisossomal

Genética • Herança autossômica recessiva • Judeus asquenazins 1: 1. 000 Htz 1: 18 • >350 casos publicados • Heterogeneidade • Gene 1 q 21 -31 • Mutações – 44 tipos – Mais comuns: N 370 S; L 444 P; 84 ins. G; IVS 2

Defeito metabólico • Deficiência de glicocerebrosidase – beta-glicosidase lisossomal Célula RE

-galactosidase fuc-gal-NAglc-gal-ceramídio fuc-gal-Naglc-gal-glc-ceramídio -galactosaminidase fuc-gal-Naglc-gal-glc-ceramídio -fucosidase gal-Naglc-gal-ceramídio -galactosidase Nacgal-gal-glc-ceramidio -hexosaminidase NAcglc-gal-glc-ceramidio -hexosaminidase -galactosidase Gal-glc-ceramidio gal-glc-ceramidio -galactosidase glc-ceramidio -glicosidase ceramídio PC-ceramídio esfingomielinase Esfingosina + Ácido graxo ceramidase

Características clínicas acúmulo MO Fígado Baço Pulmão Rins Pancitopenia Hepatoesplenomegalia Doença infiltrativa pulmonar Glomerulonefrite
 • • Necrose asséptica da cabeça do fêmur Fratura patológica Dor")
Sistema osteoarticular (20%) • • Necrose asséptica da cabeça do fêmur Fratura patológica Dor óssea Infarto ósseo Osteopenia Lesões líticas Compressão vertebral Osteonecrose
 Infantil Neuropática aguda Juvenil Hepatoesplenomegalia Lesões")
Tipo III Adulto Não neuropático Mais comum (99%) Infantil Neuropática aguda Juvenil Hepatoesplenomegalia Lesões osteolíticas Pancitopenia Deficit crescimento Retardo DNPM Aumento de tônus Organomegalia Estrabismo Óbito com 2 anos - pneumopatia Intermediária entre I e II Ataxia, convulsão, mioclonia, demência Óbito com 10 -15 anos

Laboratório • Biologia molecular • Diagnóstico pré-natal • Ensaio enzimático: betaglicosidase – 15% tipo I – 2, 3% tipo II e III

Tratamento • Reposição enzimática EV – 30 -60 ui/Kg/sem • Terapia de redução de substrato VO


Mucopolissacaridoses • Deficiência de enzima lisossômica envolvida na degradação de MPS- GAG, dermatan sulfato, queratan sulfato • Herança • Diagnóstico pré-natal • Tratamento • Diagnóstico diferencial

Quadro clínico Face infiltrada Cabeça larga Hisurtismo Sinofre Ponte nasal baixa Narinas antevertidas Bochechas proeminentes Lábios carnosos Língua larga Hiperplasia de gengiva Boca entreaberta Mandíbula pequena

• • • Baixa estatura Pele seca, amarelada Tórax curto, em barril Abdome protuso Hepatoesplenomegalia Hérnia umbilical/inguinal • Dedos curtos
 – Hurler-Schie (intermediária) – Schie (leve) •")
MPS I • Tipos – Hurler (grave) – Hurler-Schie (intermediária) – Schie (leve) • Dismorfismos, alterações esqueléticas, opacidade corneana, retardo mental (apenas Hurler) • HS, DS • Alfa-iduronidase • 22 pter-q 11 • Tratamento
 • Anomalias esqueléticas")
MPS II • Tipos – Hunter A e B (início tardio/precoce) • Anomalias esqueléticas moderadas, sem opacidade corneana, retardo mental apenas na B • HS e DS • Iduronato sulfatase • Xq 26 -27 • Tratamento

MPS III • Tipos – Sanfilippo A, B, C, D • Dismorfismos leve, anomalias esqueléticas mínimas, sem opacidade corneana, retardo mental presente • HS • Heparan-N-sulfatase (A), alfa-N-acetilglicosaminidase (B), acetil-Co. A-alfaglicosaminidase-N-acetiltransferase (C), alfa-Nacetilglicosaminidase-6 -sulfatase

MPS IV • Tipos – Morquio A e B • Dismorfismos graves, anomalias esqueléticas graves, opacidade corneana grave, retardo mental ausente • KS • Galactosaminidase-6 -sulfato-sulfatase (A), betagalactosidase (B) • 3 p 21 -q 21
 a")
MPS VI • Tipos – Maroteaux-Lamy A e B • Dismorfismos moderado (A) a grave (B), anmalias esqueléticas moderado (A) a grave (B), opacidade corneana, retardo mental (B) • DS • Arilsulfatase B • 5 p 11 -q 13

MPS VII • Tipo – Sly • Dismorfismos e anomalias esqueléticas graves, sem opacidade corneana, retardo mental tardio • DS/HS • Beta-glicuronidase • 7 cen-q 22

Oftalmologia • Opacidade corneana • Complicação – glaucoma Sistema cardiopulmonar • Insuficiência cardíaca • Infiltração pulmonar

Otorrino/fonoaudiologia • • Obstrução nasal crônica Respiração ruidosa Hipertrofia de adenóides Sialorréia Redução do reflexo de engasgo Voz grave Dificuldade de pronunciar alguns fonemas Apnéia do sono

Odontologia • • • Dentes esparsos Incisivos cônicos Mordida aberta Atraso de erupção dentária Dilaceração de raízes Destruição óssea com imagem de cistos no 1º molar e 1 -2º molar mandibular permanente • Hiperplasia alveolar • Limitação da articulação temporomandibular

Sistema musculoesquelético • • Baixa estatura Pescoço curto Subluxação C 1 -C 2 Pectus carinatum/excavatu m • Gibosidade/cifose lombar/dorsal • Limitação articular (mão em garra) • Alargamento punhos

Anomalias radiológicas • • • Cabeça grande/deformada Esfenóide plano Sela túrcica em J (cisto aracnoideo) Fechamento prematuro das suturas sagital e lambdoide Base do crânio e órbitas denso Órbitas proeminentes Ligamento estilóide calcificado (25%) Costelas alargadas Corpos vertebrais biconvexos, displásicos Porção basilar do ilíaco pouco desenvolvida Diafise de ossos longos distorcida com deformação de epífises

D. Pompe • • • Hipotonia Atraso de desenvolvimento Posturas anormais Intolerância ao exercício Infecção de repetição Broncoaspiração Cefaléia matinal Dificuldade respiratória Apnéia do sono Sonolência Perda progressiva da função pulmonar • Fraqueza muscular – dificuldade para tussir • Insuficiência respiratória progressiva – Retenção crônica de p. CO 2, redução de p O 2 e sat. O 2 – Piora durante o sono

Progressão gradual dos sintomas –D. Fabry Doença cardíaca Doença SNC Doença renal Acroparestesia 20 anos 40 anos

Sintomas – D. Fabry A D U L T O Insuficiência renal Complicação neurológica Doença cerebrovascular Disfunção cardíaca Perda da audição A D O L E S C E N T E Distúrbios gastrointestinais Angioqueratomas Fadiga C R I A N Ç A Crises de dor, acroparestesias Hipohidrose Opacidade de córnea e cristalino Febre recorrente Intolerância ao calor/frio Alterações psicossociais
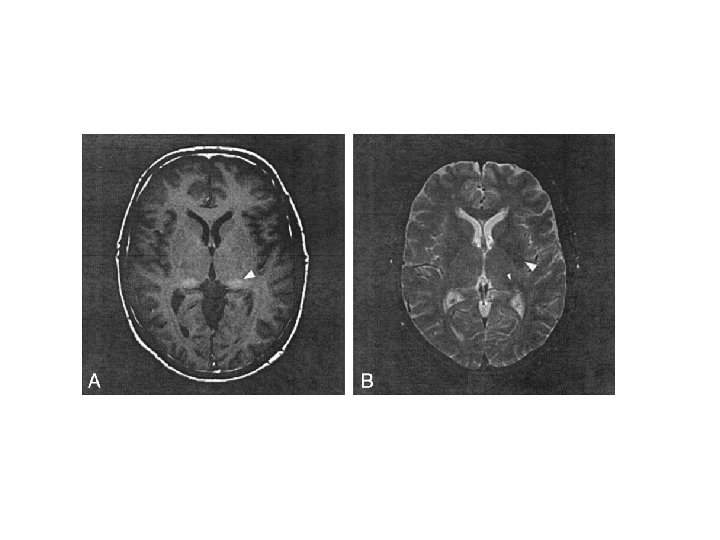
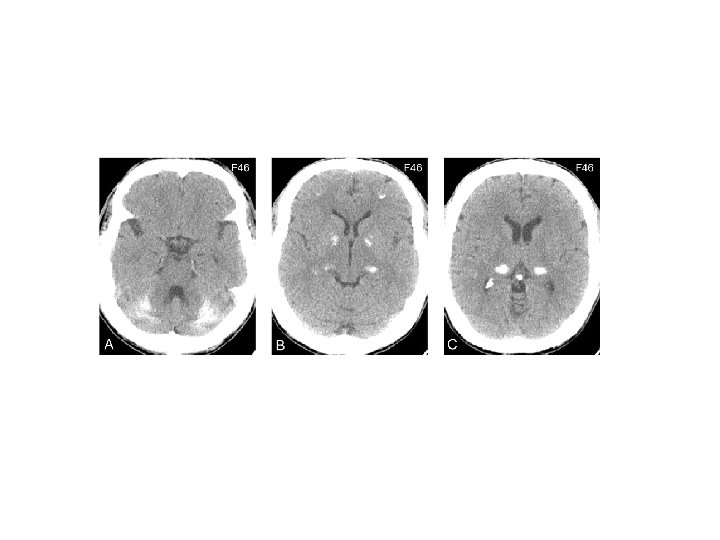

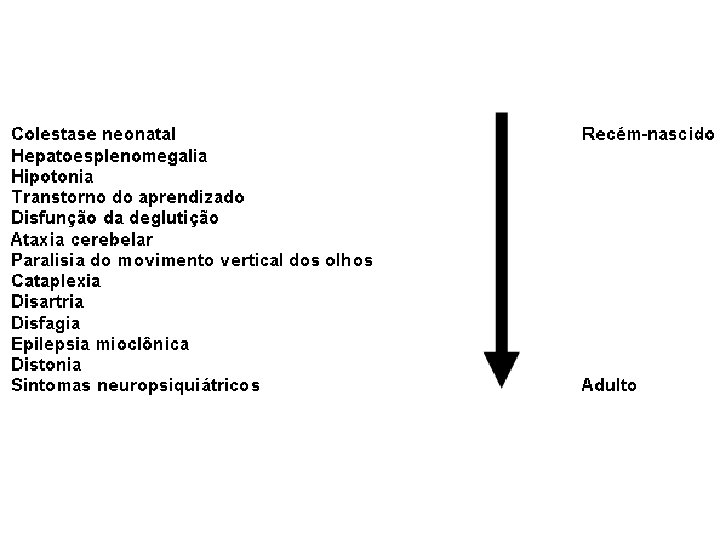
D. Niemann – Pick C Hepato/espleno Ausente=15% Idade de início variável Regride com idade Aparece antes dos sinais neurológicos Ascite fetal hidropsia Letal no período neonatal (hepato) esplenomegalia 1 a 2 a 3 a 6 a Infantil precoce 10 a 20 a juvenil Infantil tardio Paralisia do olhar vertical 30 a 60 adulto a

HIDROPSIA FETAL NÃO-IMUNE

D. Niemann-Pick C Paralisia do olhar vertical Infantil precoce Hipotonia a. DNPM Infantil tardio Alteração da marcha Atraso de fala cataplexia Juvenil Atraaso escolar Ataxia Convulsão Cataplexia
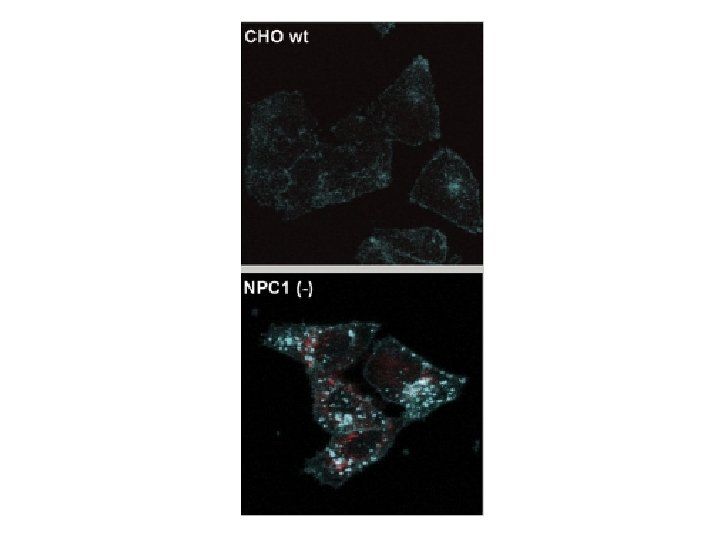

Atrofia cerebelar

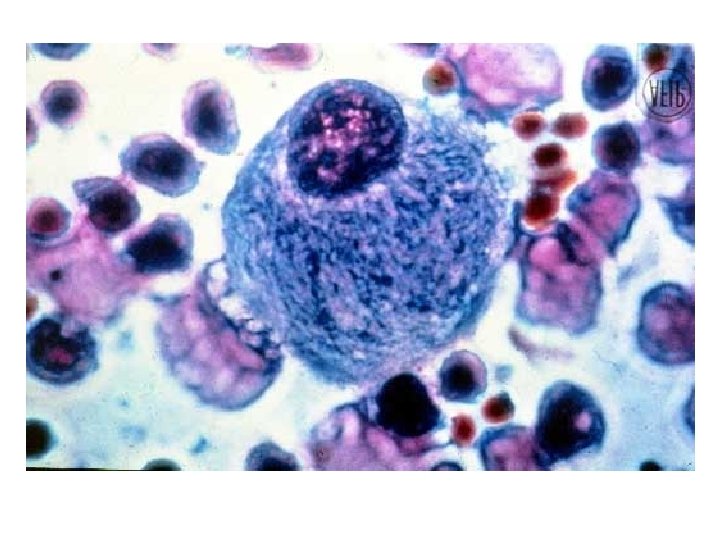
“Sea blue histiocytes” Células espumosas

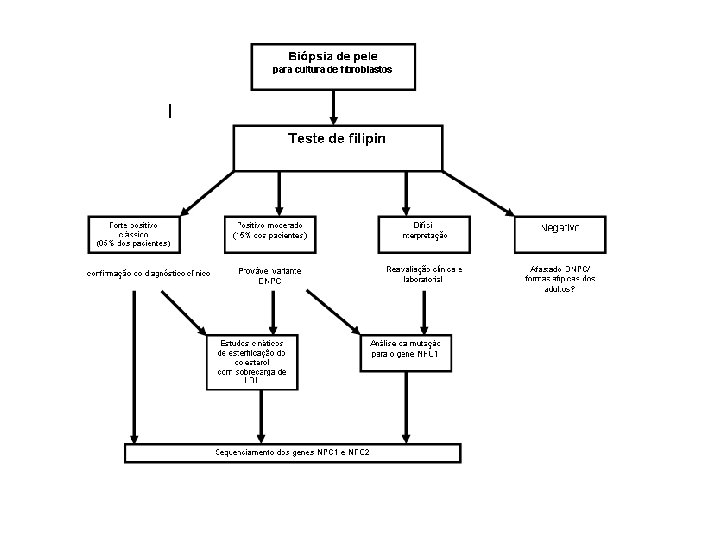
Teste de Filipin

Sequenciamento Genético • Padrão ouro para detectar mutaçoes genéticas • Maior fator limitante é o tamanho da sequencia genética a ser decodificado • Há dois genes envolvidos em NPC: NPC 1 (25 exons) e NPC 2 (5 exons) TAGCCATCGGAACGTACTCAATGATGTCGA
- Slides: 71