GENETK HASTALIKLAR II Normal karyotip ve sitogenetik tan











 Kromozom anomalileri")


















 HIV dahil enfeksiyon hastalıklarının tanısı • 5) transplantasyon, bablık testi ve")





- Slides: 39
GENETİK HASTALIKLAR II
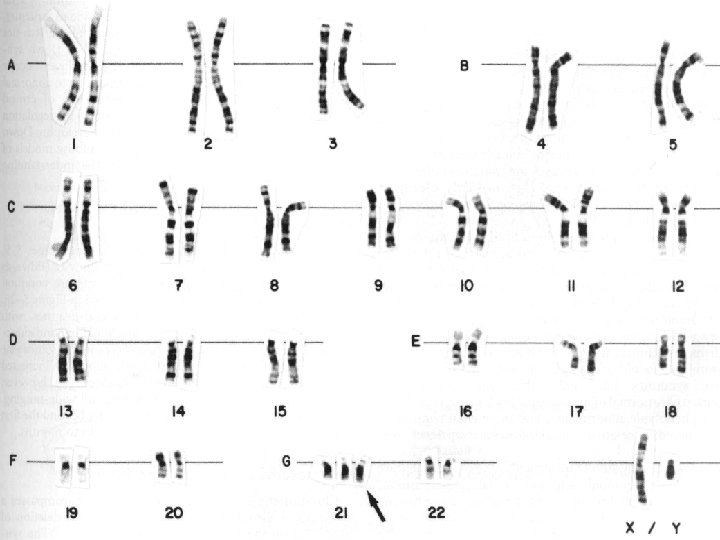
• Normal karyotip ve sitogenetik tanı yöntemleri • Normal insan karyotipi, 22 çift otozom ve 2 seks kromozomundan oluşur; 44+xx veya 44+xy • Karyotipleme yani kromozomların incelenmesi, sitogenetiğin temelidir. Kültür ortamında bölünen hücreler, metafazda kolsemid kullanılarak durdurulur; kromozomlar boyanır ve boylarına göre çiftler sıralanır. • G-bantlama=Giemsa boyası ile 400 -800 arası bant seçilebilir. • Son yıllarda profaz bantları ile, daha da fazla bant seçilir hale gelmiştir.
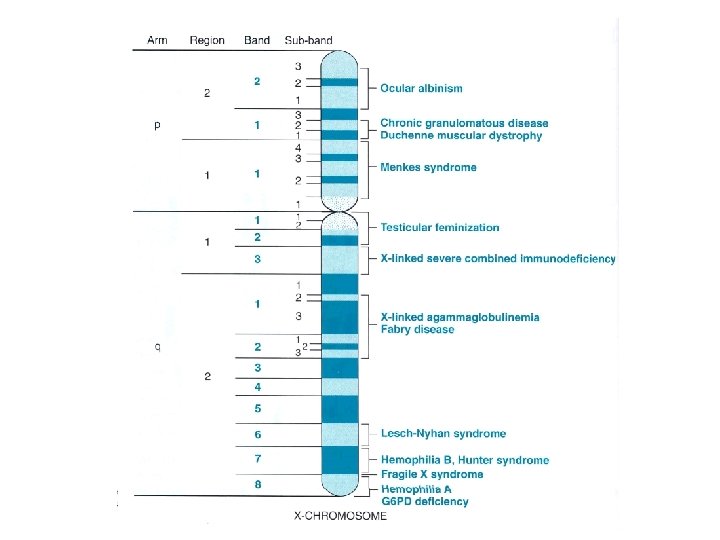
• Kromozomların İsimlendirilmesi • Kromozomun numarası daha sonra uzun ya da kısa kol belirtilir. • Xp 21. 2 dendiğinde, X kromozomunun kısa kolu anlaşılır. • p: “petit” kısa; q da ondan sonra gelen harf uzun kolu gösterir. Daha sonraki sayılar bölge, bant ve subbandı gösterir.
• Sitogenetik analiz amacıyla örnekleme • Çeşitli dokularda yapılabilir. Amniyon sıvısı, koryon vilusu, fetal kan, periferik kan, deri fibroblastları, kemik iliği, solid tm. lerde örnekleme yapılabilir. • Amniyon sıvısı: Gebeliğin 14. haftasından sonra sıvı alınır. Komplikasyon oranı %0. 5’den azdır. • Koriyon villus örneklemesi: 8 -12. haftada örnek almak mümkün. • Fetal kan örneklemesi: 17. gestasyon haftasından itibaren göbek kordonundan kan alınır. En güvenilir sonuç, ancak komplikasyon daha fazla. • Periferik kan: kolay ve en fazla kabul gören yöntem; Down sd. Ve hematolojik malignitelerde
• İnterfaz Sitogenetiği • Yakın geçmişe kadar kromozom çalışması için tek araç karyotipleme idi; dezavantajı: sadece bölünen hücrelere uygulanabilir olması. Son yıllarda başka bir teknikte, DNA probları kullanılmaktadır. Bu yöntem FISH: floresan in situ hibridizasyon tekniği; kromozomların belirli bölgelerine uyan problar floresan boyalarla işaretlenir, DNA probu spesifik hedef kromozoma bağlanır o bölge görünür hale gelir.
• Sitogenetik hastalıklar • Kromozom sayısında anormallik veya • Bir veya birden fazla kromozomda yapısal anormallik söz konusudur. • Euploid: haploid sayının tam katları • Aneuploid: 23’ün tam katları olmayan sayıda (mitoz veya meioz hatası sonucu) • Aneuploidinin en sık nedenleri: “non-disjunction” ve “anaphase lag” • Non-disjunction: 1. meiotik bölünme sırasında homolog çiftlerin ayrılmaması veya 2. meiotik bölünme veya somatik bölünme sırasında 2 kromatidin ayrılmaması= sonuçta 2 aneuploid hücre oluşur.
• Anaphase lag: meioz sırasında bir homolog kromozom veya mitoz sırasında bir kromatidin hücre çekirdeği dışında kalması • Sonuçta 1 normal ve 1 monozomik hücre ortaya çıkar • Seks kromozomlarını ilgilendiren monozomi veya trizomi: yaşamla uyumlu, ancak fenotipik anomaliler var • Otozomları ilgilendiren monozomi: yaşamla uyumlu değil, genellikle embriyogenez olmaz • Otozomları ilgilendiren trizomilerden bazıları yaşamla uyumlu
• Mozaiklik: Erken gelişme aşamalarında meydana gelen mitotik hatalar sonucunda, aynı insanda 2 veya daha fazla hücre popülasyonu ortaya çıkar. • Seks kromozomlarını ilgilendiren mozaiklik, göreceli olarak çok. • 45 X ve 47 XXX ortaya çıkmışsa, bu hücrelerden ortaya çıkan tüm hücrelerde de aynı özellikte 2 popülasyon görülecektir. Hata biraz daha geç bir bölünmede ortaya çıkarsa, 45 X/45 XX/47 XXX gibi 3 populasyon ortaya çıkar.
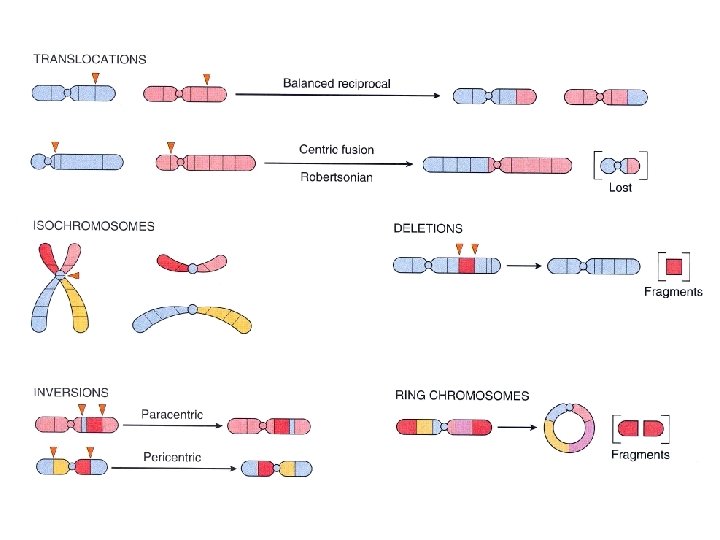
• Yapısal Anomaliler • Mevcut bantlama teknikleri ile görülebilir olması için, önemli miktarda DNA ‘yı ilgilendiren (yaklaşık 4 milyon baz çifti) bir değişiklik olmalıdır. Çevresel faktörler (mutajenler, kimyasallar, iyonize radyasyon) bu hata oranını arttırır. • Bazı hastalıklarda, kromozom kırılmalarına eğilim vardır: Fanconi anemisi, Bloom send. , Ataksitelanjiektazi; kanser riskinde artış vardır. • Delesyon: kromozomun bir kısmının kaybolmasıdır. • 46 xy, 16 p- dendiğinde, 16. kromozom kısa kolunda kayıp vardır.
• Translokasyon: Bir kromozomdan bir segment, bir diğer kromozoma taşınır. Balanced resiprokal translokasyonda, dengeli karşılıklı bir translokasyon vardır. • Balanced resiprokal translokasyon; söz konusu her iki kromozomda birer kırılma noktası ve kopan segmentlerin kromozomlar arasında yer değiştirmesi söz konusudur. • 46 xy t (2; 5) (q 31, p 14)= 2 kromozomun uzun kolu ile 5. kromozomun kısa kolu arasında karşılıklı bir translokasyon vardır. Genetik materyel kaybı yoktur; kişi fenotipik olarak normaldir. • İzokromozom: Bir kromozomun bir kolunun kaybı ve diğer kolunun duplikasyonu; kromozom ya iki kısa, ya da iki uzun koldan oluşur. Xq (kısa kolda monozomi, uzun kolda trizomi ortaya çıkar)
• Ring kromozom: özel bir tip delesyondur; kromozomun her iki ucundan birer parça kopar ve kalan kısmın uçları birleşir. Örnek: 46 xy, r (14) • İnversiyon: Bir kromozomda iki kırılmanın olması ve sonra kopan kısmın inversiyon ile tekrar yerleşmesidir • Zigotların %7. 5’inde bir kromozomal anomali olduğu tahmin edilmektedir. Bunların bir çoğu yaşamla bağdaşmaz. • Abortuslarda %50, ölü-doğum ve erken post-natal ölümlerde %5 oranında kromozom anomalisi mevcuttur. • Canlı doğumlarda kromozom anomalisi %0. 5 -1 kadardır.
• • • Otozomları ilgilendiren sitogenetik hastalıklar Trizomi 21 (Down sendromu) Kromozom anomalileri arasında en çok görülenidir. Zeka geriliğinin en başta gelen nedenlerindendir. Yenidoğanda insidens 1/700 Annenin yaşı çok önemlidir; 45’in üzeri yaşlarda insidens 1/25’e çıkar. Bulgular: %80 çocukta IQ 25 -50 arasındadır. %40 hastada konjenital kalp hastalığı vardır. Akut lösemi riski yüksektir. İnfeksiyona eğilim vardır. Ort yaşam 45 civarına ulaşmıştır; bu yaştaki hastaların beyinlerinde Alzheimer benzeri değişiklikler görülür.
• Diğer trizomiler: • 8, 9, 22, 18 ve 13. kromozomlarda da trizomiler tanımlanmıştır. • Trizomi 18 -Edwards sendromu: mental gerilik, mikrognati, konj. kalp hastalıkları, renal malformasyon • Trizomi 13 -Patau sendromu: mikrosefali, mental gerilik, kalp anomalisi, umbilikal herni, renal defekt • Cri du Chat sendromu: kedi miyavlaması send. 5 p • 1 yaşına kadar ağlaması kedi miyavlamasına benzer; ağır mental gerilik, mikrosefali vardır.
• Seks kromozomlarını ilgilendiren sitogenetik hastalıklar • Otozomal olanlara oranla daha fazladır. • Fazla veya eksik kromozom durumu, otozomal olanlara göre daha iyi tolere edilir. • (Y kromozomunda az genetik materyel bulunur ve x kromozomunun rastlantısal inaktivasyonu ile açıklanır) • X kromozomu inaktivasyonu • 1961 Mary Lyon tarafından Lyon hipotezi ortaya konmuş. • Lyon hipotezi: X kromozomlarından sadece 1 tanesi genetik olarak aktiftir. • Diğer X kromozomu (anne veya baba kaynaklı) heteropiknoza uğrar ve inaktiftir.
• X inaktivasyonu embriyonik yaşamın 16. gününde blastositin tüm hücrelerinde rastlantısal olarak gerçekleşir. • Aynı X kromozomunun inaktif hali, anne hücreninki ile aynı şekilde yavru hücrelerde devam eder. • İnaktif X: interfaz çekirdeğinde Barr cisimciği veya X kromatini; kadınların tüm somatik hücrelerinde görülür. • En kolay bukkal smear’de skuamöz epitel hücrelerinde görülür. • İlk zamanlar, inaktif X’in tüm genlerinin suskun olduğu sanılmaktaydı. Ancak son çalışmalar, birçok genin X inaktivasyonu dışında kaldığını göstermektedir.
• Y kromozomu: erkek cinsiyet gelişimi için gerekli ve yeterli • X kromozomu sayısından bağımsız olarak tek bir Y varlığı, erkek cinsiyeti belirler. • Tüm seks kromozomu hastalıklarında ortak noktalar • Genel olarak belirsiz, seksüel gelişme ve fertilite ilgili problemlere yol açarlar. • Çoğunun tanısı, doğumda zordur. Genellikle puberte sonrasında tanınırlar. • Genel olarak erkek veya kadında, X kromozomu ne kadar fazlaysa, mental retardasyon şansı o kadar artar.
• • Klinefelter sendromu Erkekte hipogonadizm: 2 veya daha fazla sayıda X kromozomu ve 1 veya daha fazla sayıda Y kromozomu • Seks kromozomlarıyla ilgili hastalıklar arasında en sık olanlardan biridir. • Erkek hipogonadizminin en sık nedenlerinden biridir. • %82 47 XXY klasik Klinefelter-meiotik nondisjunction • %15 mozaiklik 46 XY/47 XXY; 47 XXY/48 XXXY • X sayısı arttıkça, anomaliler artar.
• Bulgular: • Uzun boylu görünüm, küçük atrofik testisler, küçük penis • Sekonder erkek seks karakteristiklerinin yokluğu (ses, kıllanma) • IQ hafif düşük • Testiste: seminifer tubuluslar atrofiktir • Meme ca normalin 20 katı fazla görülür; ekstragonadal germ hücreli tm, SLE görülebilir. • XYY sendromu • 47 XYY veya daha fazla Y: aşırı uzun boy akneye eğilim • Fenotip genellikle normaldir
• • • Turner sendromu Komplet veya parsiyel X kromozomu monozomisi Primer amenore vakalarının en önemli nedenidir; 1/3’ü %57 hastada tam bir X yokluğu: 45 X %43 diğer anomaliler; kısa kol delesyonu; kısa veya uzun koldan bölümlerin delesyonu ve ring kromozom oluşumu; mozaiklik gösteren vakalar • 45 X Turner sendromu vakalarında anomaliler ağır: tanı doğumda veya erken çocukluk çağında konabilir. • Kısa boy, aort koarktasyonu, infertilite, amenore, doğumda periferik lenfödem
• Multi-X kadınlar • Büyük çoğunluğu normal • Ancak X fazlalaştıkça, mental retardasyon eğilimi artar • Hermafroditizm ve Psödohermafroditizm • Gerçek hermafrodit: Over ve testis dokusunun varlığıçok nadir • Psödohermafrodit: fenotipik ve gonadal seks arasında uyumsuzluk • Dişi psödohermafrodit: over ve erkek dış genital organlar; konjenital adrenal hiperplazi en önemli neden • Erkek psödohermafrodit: testis ve dişi dış genital organlar; androjen sentez veya etkisinde bozukluk var
Klasik kalıtım uğramayan tek gen hastalıkları Bazı tek gen hastalıklarının, klasik Mendel Kanunlarına uymadığı bilinmektedir. Bunlar 4 bölüme ayrılır. – – Üçlü nukleotit tekrar mutasyonları Mitokondriyal genlerdeki mutasyonlar Genomik imprinting Gonadal mozaiklik
• Üçlü tekrar mutasyonları • Frajil X sendromu • 3 nukleotidli uzun tekrarlayan sekans ile karakterizedir • Frajil X sendromu, genetik nedenli mental gerilikler içinde Down sendromundan sonra 2. sıradadır • X’e bağlı bir hastalıktır • FMR-1 gen mutasyonu vardır; x uzun kolunda devamlılık bozulmuştur •
• • IQ 20 -60 arasındadır Uzun yüz, büyük mandibula, büyük testisler Yüksek damak, mitral valv prolapsusu Aşırı ekstansiyona gelen eklemler • Erkek hastalığa yakalanır; X’e bağlı hastalıklardaki gibi • Ancak bazı geçiş özellikleri x’e bağlı res. Geçiş paternine uymaz: • Bu mutasyonu taşıyan erkeklerin %20’si hasta değildir • %50 taşıyıcı kadın mental retardedir
• Başlıca trinukleotid tekrar bozuklukları • Frajil X sendromu CGG • Friedreich ataksisi GAA • Myotonik distrofi CTG • Huntington hastalığı • Spinoserebellar ataksinin çeşitli tipleri
• Mitokondriyal genlerdeki mutasyonlar Leber herediter optik nöropati Genlerin çoğu çekirdekteki kromozomlarda yer alır. Bu genler, Mendel kanunlarına uyarak kalıtıma uğrar. Oysa bazı genler bu kurallara uymaz. İnsan mitokondriyal DNA ‘sı 37 gen içerir; bunların 13’ü oksidatif fosforilasyonda görevli enzimleri kodlar. Bozukluklarında da, primer olarak oksidatif fosforilasyona bağımlı organlarda hastalık ortaya çıkar: santral sinir sistemi, çizgili kas ve kalp kası, karaciğer, böbrek
• Mitokondriyal kalıtımla ilişkili hastalık azdır, olanların da çoğu nöromuskuler sistemi etkiler • Leber herediter optik nöropatisinde, 15 -35 yaş arasında görme bozukluğu ortaya çıkar. Ovumda bol mitokondri vardır; spermatozoada ise varsa bile çok az. Anneden hastalık kızı ve oğluna geçer, fakat sadece kız çocuk kendi kızlarına hastalığı geçirebilir.
• Genomik imprinting • Her genin 2 kopyası, anne ve babadan çocuğa geçer. • Anne ve babadan geçen bu genlerin fonksiyonel olarak birbirinden farkı olmadığı düşünülmekteyken anlaşılmış ki, bazı genlerde bir fark var: anne veya babadan geçen kopyanın birisi bazen inaktive olabiliyor: genomik imprinting • Ovum veya spermde olur, mitozla tüm hücrelere geçer • Bu olaya örnek iki hastalık var • Prader Willi sendromu ve • Angelman sendromu
• • Prader Willi sendromu Mental retardasyon Kısa boy Hipotoni Obezite Küçük el ve ayak Hipogonadizm Babadan gelen 15. kromozomun q 12 bandında delesyon vardır • Angelman sendromunda ise, aynı bölgede anneden gelen kromozomda delesyon vardır: mental retardasyon, ataksi, konvülziyon ve anormal gülme
• Gonadal mozaiklik • Her otozomal hastalıkta bazı ebeveynler hasta olmaz; bu yeni mutasyonla açıklanabilir • Ancak böyle bir durumda da, sadece mutasyon olan çocuk hastadır, kardeşleri hasta olmaz • Oysa bazı otoz. dom. hastalıklarda (osteog. imperfekta) normal anne babaya rağmen birden fazla hasta çocuk vardır; gonadal mozaiklik ile açıklanmaktadır. • Gonadal mozaiklik, erken gelişim (embriyonik) sırasında postzigotik mutasyon sonucu olur.
• Moleküler Tanı • İnsan genomu projesinin tamamlanmasıyla, DNA probları genetik ve edinsel hastalıkların tanısı için önemli bir yardımcı oldular. • Moleküler tanı yöntemlerinin kullanıldığı durumlar • 1) genetik hastalıkların temelindeki kalıtsal mutasyonların saptanmasında prenatal veya doğumdan sonra • 2) tümörlere neden olan edinsel mutasyonların saptanması • 3) özellikle hematopoetik sistem kaynaklı tümörlerin doğru tanısı ve sınıflandırılması
• 4) HIV dahil enfeksiyon hastalıklarının tanısı • 5) transplantasyon, bablık testi ve adli tıpta doku gruplarının saptanması • 6) bunlara ek olarak: akciğer ca. ’da olduğu gibi, hastalığa duyarlılığı etkileyen polimorfizmin saptanması (P-450 monooksigenaz sistemindeki bilinen bir polimorfizm, sigaraya bağlı akciğer ca. ’ya eğilimi arttırır) • Hedef: komple genetik analiz ile muhtemel genetik hastalıklar ve çevre etkisiyle olan hastalıklar açısından riski saptamak mümkün olacak • Genetik bilgiyi kullanırken gizlilik ilkesi ve etik önemli
• Genetik hastalıkların tanısı, genetik materyelin incelenmesi ile mümkün olur. • Başlıca 2 yöntem kullanılmaktadır 1) Sitogenetik analiz/karyotipleme 2) Moleküler analiz Prenatal kromozom analizi hangi durumlarda yapılır? anne yaşı >34 ise trizomi riski artar anne veya baba translokasyon ya da inversiyon taşıyıcısı ise önceki çocukta kromozom anomalisi varsa anne/baba X’e bağlı genetik hast. taşıyıcısı ise
• • • Postnatal kromozom analizi Periferal kandan lenfositler alınarak yapılır Multipl doğumsal anomali Açıklanamayan mental gerilik veya gelişimsel gecikme Şüpheli anöploidi-özellikle Down açısından Şüpheli dengelenmemiş otozom-Prader Willi Şüpheli seks kromozom anomalisi-Turner sendromu Şüpheli frajil X sendromu İnfertilite-seks kromozom anomalisini ekarte etmek için • Multip spontan abortus-anne/babanın taşıyıcılığını ekarte etmek için
• Birçok genetik hastalık, karyotipleme ile saptanamayacak düzeyde küçük değişiklikler sonucu meydana gelir • Geleneksel olarak tek gen hastalıklarının tanısı: • Anormal gen ürünleri (mutant Hg veya enzim) • Ya da bunun sonucunda ortaya çıkan klinik etkilerin (anemi, mental gerilik) saptanması ile yapılırken, • bugün doğrudan DNA düzeyinde patolojiyi göstermek mümkün
• Rekombine DNA teknolojisinin önemli avantajları: • Duyarlı bir teknik olması • PCR (polimeraz chain reaction) kullanımı sayesinde, tek bir hücreden alınan örnek bile çoğaltılarak tanıya gidilebilir • Beyinde bir patoloji varsa, bu patoloji sonucu oluşan anormal madde veya enzim de beyin dokusundan saptanabilir; oysa moleküler inceleme tüm hücreler aynı DNA’yı içerdiği için, her hücrede yapılabilir • Direkt gen tanısı -PCR ve RT-PCR ile • İndirekt gen tanısı- bir hastalıkta çeşitli mut. varsa • Bu yöntem daha uygun-linkage analiz kullanılır