Genetics Down Syndrome Down Syndrome Is a genetic


 (ie, presence")





 crease, wide gap")


: 47 XX +21 or 47, XY=21 Translocation")










 shows that")






 karyotype")


- Slides: 36
Genetics
Down Syndrome
Down Syndrome Is a genetic disorder caused by extra genetic material (DNA) (ie, presence of an extra 21 st chromosome) Common autosomal aneuplody (trisomy 21). It is the common of the chromosomal disorders and leading cause of mental retardation. Incidence : 1 in 700 births and the incidence increases with the advancing age of the mother usually above 35 yrs.
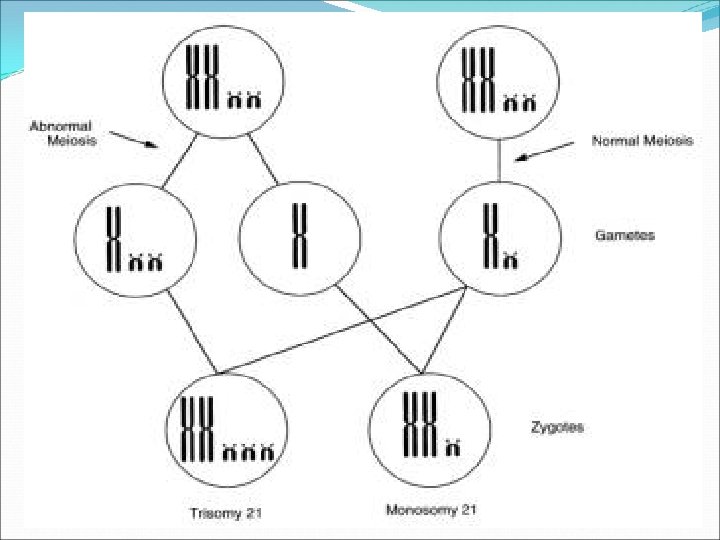
Cont… Human cells normally have 46 chromosomes that can be arranged in 23 pairs. One set of 23 chromosomes comes from the mother (ovum) and the other half of the 23 pairs comes from the father (sperm cell). Trisomy 21 In Down syndrome, 95% of all cases are caused by either the sperm or the egg cell having two 21 st chromosomes instead of one, so the resulting fertilized egg has three 21 st chromosomes. Hence the scientific name, trisomy 21. Recent research has shown that in these cases, approximately 90% of the time the abnormal cells are the ovum. The cause of the extra chromosome isn't known, but there is definitely connection with the mother's age.
Robertsonian translocation Occurs in 3 -4% of cases. In this case, the genetic material is rearranged so that some of the 13 th, 14 th, or 15 th chromosome is replaced by an extra copy of genetic material from the 21 st chromosome. The overall number of chromosomes remains normal (46 chromosomes in 23 pairs), but there are 3 copies of the 21 st chromosome material. Partial trisomy 21 Sometimes the extra genetic material only comes from part of the long arm of the 21 st chromosome (21 q), and this is called partial trisomy 21
Mosaicism and trisomy 21 In these cases, people have a mixture of cells. Some cells have a normal set of chromosomes, and other cells have trisomy 21. In cellular mosaicism, the mixture is seen in different cells of the same type. In tissue mosaicism, one set of cells, such as all blood cells, may have normal chromosomes, and another type, such as all skin cells, may have trisomy 21.
Genes that may have input into Down syndrome include: SOD 1 (superoxide dismutase 1 gene) overexpression may cause premature aging and decreased function of the immune system. COL 6 A 1 (alpha-1 collagen VI gene) overexpression may be the cause of heart defects. ETS 2 (ETS 2 oncogene) overexpression may be the cause of skeletal abnormalities. CAF 1 A (chromatin assembly factor 1, p 60 subunit) overexpression may cause problems with DNA synthesis. CBS (cystathione beta synthase) overexpression may disrupt metabolism and DNA repair. DYRK 1 A (dual-specificity tyrosine phosphorylation-regulated kinase 1 A) overexpression may be the cause of mental retardation. CRYA 1 (alpha-1 crystallin) overexpression may be the cause of cataracts. GART (glycinamide ribonucleotide synthetase) overexpression may disrupt DNA synthesis and repair. IFNAR (interferon alpha receptor) overexpression may interfere with the immune system as well as other organ systems. Remember that no gene has yet been fully linked to any feature associated with Down syndrome.
Clinical features A. General Mental retardation, hypotonia (poor Moro’s reflex and hyper flexibility of the joint ), reduce life expectancy by 40%. B. Face and skull: Oblique palpebral fissure, marked epicanthal fold, flat nasal bridge , low set ears, and flat occiput, protruding tongue. C. Thorax and abdomen: congenital heart malformation (ventricular septal defect), duodenal atresia , imperforated anus, umbilical hernia.
D. Hand feets: short and broad hands , single palmar (simian) crease, wide gap between 1 st and 2 nd toes (sandal gap) , clinodactyl (hypoplasia of the middle phalanx with a single flexion crease of the 5 th finger.
Characteristic flat facies with hypertelorism, depressed nasal bridge, protrusion of the tongue, a single palmar simian crease Small auricle and anomalies of the folds
wide gap between the first and second toes protuberant abdomen and an umbilical hernia
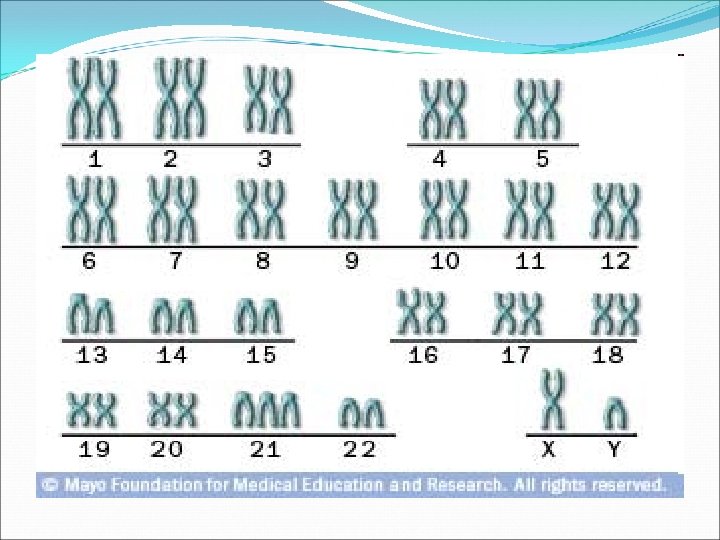
Investigation 1. Karyotyping Trisomy 21 (about 95%): 47 XX +21 or 47, XY=21 Translocation 21/14, 21/22
Treatment 1. Advice to the parents. 2. Genetic counselling. i. Antenatal diagnosis is possible by aminocentesis (15 -16 wks) and choronic villi sampling (9 -11 wks) ii. Detection of carrier is possible by chromosome analysis, DNA analysis technique (thalassaemia, haemophilia)
Supportive management of the morbid condition such as: Recurrent chest infection Appropriate management of the congenital anomalies such as imperforated anus , VSD
Down Syndrome Life Expectancy Statistics: Down Syndrome life expectancy in utero is only a few weeks / months. Three quarters of babies with Down Syndrome will die in utero. Down Syndrome life expectancy for babies and infants, sees approximately 15% dieing before one year of age. Down Syndrome life expectancy for the next 35% will be death before 50 years of age. Down Syndrome life expectancy for the remaining 50% is more than 50 years of age.
Main causes of shortened Life Expectancy During first year of life: First and foremost, heart problems. Second, problems with the digestive system : 2 a. Esophageal atresia 2 b. Tran esophageal fistula 2 c. Hirsch sprung disease 2 d. Duodenal atresia
Cytogenetic disorders involving sex chromosomes : Two factors are peculiar to the sex chromosomes : - 1. Lyonization or inactivation of all but one X chromosomes and 2. The modest amount of genetic material carried by the Y chromosomes
Lyon hypothesis 1. Only one of the X chromosomes is genetically active 2. The other X of either maternal or paternal origin undergoes heteropyknosis (condensed) and is rendered inactive 3. Inactivation of either maternal or paternal X occurs about the 16 th day of embryonic life and 4. Inactivation of the same X chromosomes persists in all the cells derived from each precursor cell.
Cont. . *** Recent studies shows that many genes escape X inactivation. At least some of the genes that are expressed from both X chromosomes are important for normal growth and development. e. g ; patients with monosomy of the X chromosomes (Turner syndrome: 45, X) have severe somatic and gonadal abnormalities. Because, if a single dose of X – linked genes were sufficient, no detrimental (harmful) effect would be expected in such cases.
Cont. . Although one X chromosome is inactivated in all cells during embryogenesis, it is selectively reactivated in all cells during oogonia before the first meiotic division. Thus, it seems that both X chromosomes are required for normal oogenesis. Y chromosomes : both necessary and sufficient for male development Presence of single Y chromosomes determines the male sex. Gene that dictates testicular development has been located on the distal short arm.
Cont… With this background, we review some features that are common to all sex chromosomes disorders : 1. Chronic problems relating to sexual development and fertility 2. Difficult to diagnose at birth, many are first recognized at the time of puberty 3. In general, the higher the number of X chromosomes, in both male and female, the greater the likelihood of mental retardation.
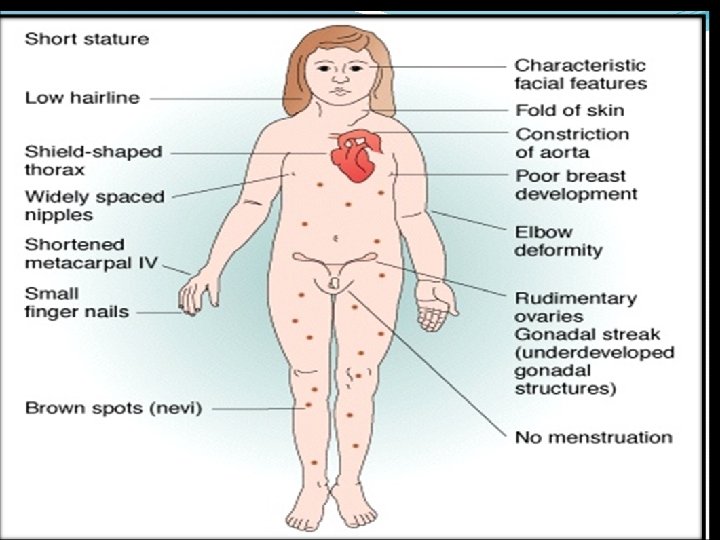
Tunner’s syndrome Results from complete or partial monosomy of the X chromosomes and is characterized primarily by hypogonadism in phenotypic females It is a sex chromosomal aneuploidy here a female has lost an X chromosome. Frequency : 1 in 2000 liveborn females Karyotype: 45 X (57%) 29% mosaics (45, X with 1 or more karyotypically normal or abnormal cells type 14 % -- structural abnormalities of X chromosomes
Cont… 99% of 45, X conceptuses are nonviable so many tests (PCR) shows that the prevalence of mosaic Turner syndrome increases to 75%. In patients who are truly 45, X orin whom the proportion of 45, X cells is high, the phenotypic changes are more severe than the mosaicism.
Clinial features The disorders may be recognized at the birth. A newborn presents with lymph edema of the dorsum of hand feet's, and loose skin folds at the nape of neck. An older children will have short stature , short webbed neck and low posterior hairline. Ears are anomalous and prominent. The palate is high arched and mandible is small. Chest is broad shield like with widely spaced hypoplastic nipples.
There is carrying angle at elbow. The fourth metacarpals and metatarsals are short. At puberty sexual maturity fails to occur. Adult stature is less than 145 cm. Associated congenital anomalies are: Kidneys : horse shoe kidneys, double or cleft renal pelvis. Heart : coarctation of aorta Ears : hearing defective Ovarian dysgenesis Intellectual development is normal.
MANAGEMENT AND TREATMENT Cardiac evaluation including blood pressure and ECHO is recommended as the base line and then every year. Height should be monitored. If the growth is not proper the growth hormone therapy is useful and may increase the final height by 8 to 10 cm. Counseling regarding the behavioral problems due to short stature , amenorrhea and sterility is the integral part of management. Yearly TSH monitoring should begin from 10 yrs of age.
Ovarian hormone replacement should be started at age of 14 yrs. To start with conjugated estrogen (0. 3 mg/dl) or ethinyl estradiol (5 -10 microg/dl) is used for 3 to 6 months ; then increased to 0. 625 to 1. 25 mg (conjugated estrogen) or 20 to 50 microg/dl (ethinyl estradiol). After 6 month to 1 year , cyclical therapy with estrogen and progesterone is started.
Regular audiometry should be done in adults or earlier if indicated. Evaluation of renal malformation by USG at first contact should be done. Prophylactic gonadectomy in tunner’s syndrome due to chances of gonadoblastoma.
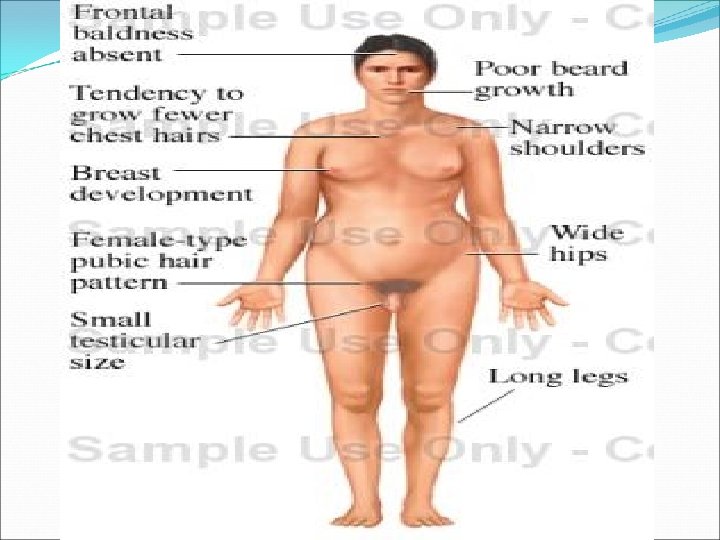
Klinefelter Syndrome Defined as male hypogonadism that occurs when there are two or more X chromosomes and one or more Y chromosomes It is numerical sex chromosomal abnormalities (aneuploidy) Here a male with extra X chromosomes. Frequency is about 1 in 500 live births. It can rarely be diagnosed before puberty, because the testicular abnormality does not develop before early puberyt.
Cont… Classic pattern of Klinefelter syndrome is associated with a 47, XXY (82%) karyotype This results from the nondisjunction during the meiotic division in one of the parents. Maternal nondisjunction at the first meiotic division is more common than paternal first meiotic division. There is no phenotypic difference between those who receive the extra X chromosomes from their father or from mother.
Clinical features Small testis Poorly developed secondary sexual characteristics –the growth of the pubic and the facial hair is often late. Gynecomastia Infertility May be mild reduction in verbal skill, language difficulty, attention deficit, as well as children has psychosocial , learning or school adjustment problems with hypogonadism, small testis , infertility and gynecomastia.
MANAGEMENT AND TREATMENT This includes behavioral and psychosocial rehabilitation. Testosterone therapy should be started in middle to late adolescence with monitoring of level. Testosterone enanthate is used in the dose 200 mg /day every 10 to 24 days IM(adults) In adolescents -100 mgevery 10 to 14 days.