Genes trees vs Species tree Phylogentic trees based
 Topology")
Genes trees vs Species tree Phylogentic trees (based on 1 or multiple genes) Topology and bl information on the evolutionary history of a taxon consensus vs. concatenation Gene trees ILS LGT = HGT Gene flow (common between Incongruences in topology Network graphs alternative hypotheses, reticulation S p eci es t ree…? ? ? taxa in time intervals just after speciation) Gene duplication Hybridization (successfull interbreeding between genetically divergent taxa – hybrid zone: where the hybrid offspring of two divergent taxa are prevalent and there is a cline in the genetic composition of populations from one taxon to the other) Introgression (hybridization without the formation of a new taxonomic lineage)

S p e c i e s t r e e v s. g e n e t r e e s Human-chimp-gorilla
 Genes can have unequal rates of evolution. (B) Gene")
Species tree…? ? ? A) Genes can have unequal rates of evolution. (B) Gene loss and gene duplication are common. Mitchell, M. W. & Gonder, M. K. (2013) Primate speciation: A case study of African apes. Nature Education Knowledge 4(2): 1 C) Gene flow can occur between lineages after their separation. D) Recombination between neighboring regions species phylogenies and gene histories do not match.

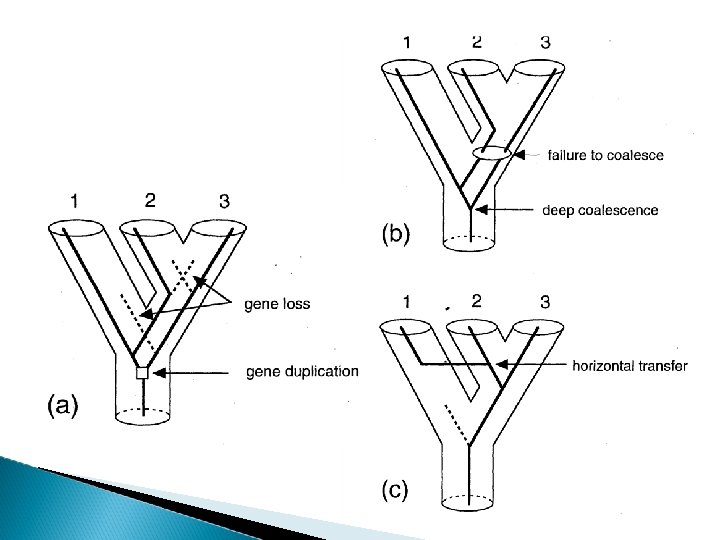
Incongruences between gene phylogenies Lineage sorting There is no guarantee that alleles sort in a lineage to match up with the overall species pattern. Speciation events gene variation in population the gene will sometime assorts with a pattern that does not match the species pattern the gene tree will be “discordant” with the species tree

http: //biologos. org/blog/evolution-basics-species-trees-gene-trees-and-incomplete-lineage-sorting
 the MRCA does not occur in the")
In case of INCOMPLETE LINEAGE SORTING (ILS) the MRCA does not occur in the most recent population in which the two lineages cooccur the coalescence of lineages from two relatively distant related species might occur more recently than that of lineages from two more closely related species!!!

Incongruences between gene phylogenies Considering populations …

Species tree…? ? ? Model of nucleotide substitution estimation of gene trees Model relating gene trees to the history of the species estimation of species trees Coalescence of gene lineages multiple loci !!! Multiple individuals sampled for one species MRCA (most recent common ancestor) with the closest related species Gene lineages of one species are more likely to share a MRCA with closely related species than with distant related species.

Species tree…? ? ? Phylogenetic history sampling strategy Multiple individuals Gene lineages Probability of coalescence (monophyletic group within a species) Species divergence history polyphyly paraphyly non-reciprocal monophyly Multiple individuals per species Multiple loci per species

polyphyly paraphyly non-reciprocal monophyly Probability of relationships


20 individuals loci similar 20 15 15 10 10 5 5 20 0 5 10 15 15 individuals, 5 loci 15 5 individuals, 15 loci 0 20 In case of recent species divergence In case of older species divergence 5 10 100 15 20 no significant gain in accuracy 75 10 50 low accurrancy high 5 Knowles (2010) 0 25 5 10 15 20 0 25 50 75 100

Species tree vs. gene trees coalescent The coalescent describes the relationship between the demographic history of a large population and the shared ancestry of individuals randomly sampled from it, as represented by a genealogical tree. constant-sized (a) and exponentially growing populations (b) Moving back in time from the present, we follow the number of lineages in the genealogy in each generation. This value decreases when two lineages share a common ancestor = coalescence event, and increases when sampled individuals are encountered (a sampling event). Probability that a coalescence event occurs at a particular time is inversely proportional to the population size at that time pattern of observed coalescence and sampling events can be used to estimate the demographic history.
 sampled from a population may")
Coalescent theory It models how gene variants (= alleles) sampled from a population may have originated from a common ancestor (CA). MSC It models genes divergence in a genealogy base on a stochastic process. ASSUMPTION: Hardy-Weinberg equilibrium (population genetics): No gene flow No natural selection No recombination Random mutation (polymorphisms!!!) ? • No gene flow ( no admixture) • No natural selection • No recombination • No mutation • Population infinitly large • No emigration & no immigration • Members able to breed and random mating
 the pattern of species descent analyzing a genetic locus for different")
Phylogenetics (phylogenetic trees) the pattern of species descent analyzing a genetic locus for different species; substitution, state changes!! Estimated gene tree species tree Genealogical methods (coalescent!) Time constrain, fix mutation rate - molecular clock! PHYLOGEOGRAPHY: The use of estimated genealogies to study geographical history and structure of populations and species. EVOLUTIONARY GENETICS EPIDEMIOLOGY
 Coalescence = find the MRCA Coalescence process: mathematical model(s) for the random")
Coalescent (1) Coalescence = find the MRCA Coalescence process: mathematical model(s) for the random joining of sampled gene lineages as they are followed back in time. A coalescence event merge two lineage into one till they root. It results from consideration of large-sample approximation of common population genetic models. • Number of sampled lineages • Population size (N)

Coalescent units: units time interval between speciation events
 Number of loci Basic assumption: assumption events that occur in one population")
Coalescent (2) Number of loci Basic assumption: assumption events that occur in one population are independent independen of what happens in other populations within the phylogeny. Lenght of the DNA sequences for each locus Proportion of informative size R-F distance (Robinson-Foulds measure of quantitative dissimilarities between two trees without relying on an evolutionary model, and is therefore well suited to address discordance caused by error; defined for unrooted and non-binary trees) Heterogeneity sequence data and R-F value high more data for resolution of species tree No missing loci/data…. is better!
 deep coalescence in the past gene tree – species tree Species tree")
Coalescent (3) deep coalescence in the past gene tree – species tree Species tree can be inferred by a combination of morphological and molecular characters, geologic or biogeographic data In simulations: Species tree Simulate genalogies genealogies Simulate DNA data estimated species tree estimated gene trees
 recent divergence coalescence ancestral species deepest split Knowles Syst. Biol. 2009")
Coalescent (4) recent divergence coalescence ancestral species deepest split Knowles Syst. Biol. 2009
 Reduced gene tree discord when time between successive speciation becomes larger long")
Coalescent (5) Reduced gene tree discord when time between successive speciation becomes larger long internal branches in the species tree, older species origins. Insufficient time for fixation of gene lineages, time between successive speciation is short (less than 4 Ne) rapid species diversification or recent species divergence gene lineages may coalesce with gene lineages not belonging to the most closely related species.
 BEST (Bayesian estimation of species trees): gene trees are generated from species")
Coalescent (6) BEST (Bayesian estimation of species trees): gene trees are generated from species trees from coalescence process, DNA data are estimated from gene tree from mutation process. Incongruence due only to (different) coalescence! Species tree coalescence Prob(G/S) inference Gene tree inference mutation Prob(D/G) DNA sequences Discrepancies between gene trees and the species tree are due exclusively to lineage sorting with free recombination between genes and no recombination within genes. sequences gene trees species tree
 BCA = Bayesian concordance analysis (Cranston et al. 2009) (the amount of")
Coalescent (7) BCA = Bayesian concordance analysis (Cranston et al. 2009) (the amount of overlap in Bayesian PP distributions of trees for different genes contains information on the degrees of concordance among genes) the dominant tree is built from the clades that are inferred to be true for a high proportion of genes in the genome. Variability in genealogies CF = concordance factors (proportion of the genome for which a given clade is true) – (Baum 2007) – measure of genomic support ILS, LGT, gene flow, recobination, introgression are accounted for!! How much of the genome (how many of the sampled genes) truly have a particular clade in their tree Statistical support (PP or bootstrap values) measure how confident we are that a particular clade truly is that one on the tree! !
 MDC = minimizing deep coalescence based on searching the species trees that")
Coalescent (8) MDC = minimizing deep coalescence based on searching the species trees that minimize the implied number of deep coalescence in the contained gene trees (parsimony based method). Gene tree: Species tree: (A, (C, (B, D))) (A, (B, (C, D)))
 Extra lineages number of gene lineages minus one Gsi, genealogical sorting index")
Coalescent (9) Extra lineages number of gene lineages minus one Gsi, genealogical sorting index 1. Is a statistic that quantifies the common ancestry of groups of individuals on a phylogenetic tree; 2. summarizes incongruence between independently inferred gene trees; 3. best calculated with larger groups and balanced group sizes, no species missing in any gene; 4. tree do not need to be fully resolved

BUCKy: combine molecular data from multiple loci estimates the dominant history of sampled individuals, and how much of the genome supports each relationship, using Bayesian concordance analysis does not assume that genes (or loci) all have the same topology detects groups of genes sharing the same tree (accounting for uncertainty in gene tree estimates), combined to gain more resolution on their common tree no assumption is made regarding the reason for discordance among gene trees BEAST: BEAST cross-platform program for Bayesian analysis of molecular sequences using MCMC; it is entirely orientated towards rooted, time-measured phylogenies inferred using strict or relaxed molecular clock models; uses MCMC to average over tree space, so that each tree is weighted proportional to its posterior probability.

STEM: inferring maximum likelihood species trees from a collection of estimated gene trees under the coalescent model; gene trees must be rooted and must satisfy the molecular clock (“relaxed” accounts for different mutation rates/ “strict” neutral mutation rate. STEM-hy (hybridization): takes as its input one gene tree for each locus; can handle different taxon samples across genes; branch lengths must be estimated subject to a molecular clock; gene trees must be fully resolved. SVDquartets: Sample four species Select one lineage at random from each species Estimate the quartet relationships among the four sampled lineages Restore the species labels (but lineage quartets are saved, too) Generate all quartets (small problems) or sample quartets (large problems) Estimate the correct quartet relationship for each sampled quartet Use a quartet assembly method to build the tree

ADAPTIVE RADIATION Rapid diversification of organisms multitude of new forms, change in the environment new resources available, new challenges, new environmental niches; generalized / specialized Darwin’s finches

Ecological opportunity promotes rapid proliferation of phylogenetic and ecological diversity. ADAPTIVE RADIATION Anolean lizards Islands vs. continental regions (more stable, less frequent ecological opportunities) recent common ancestor (RCA) SPECIATION, phenotypic adaptation (different morphological and physiological traits for diverse environments).

What defines an adaptive radiation? Macroevolutionary diversification dynamics of an exceptionally species-rich continental lizard radiation. Pincheira-Donoso D, Harvey LP, Ruta M. (2015) BMC Evol Biol density-dependent declines in diversification via niche saturation over evolutionary time adaptive radiation through niche filling

Liolaemus have diversified under a density-dependent process with slightly pronounced apparent episodic pulses of lineage accumulation, which are compatible with the expected episodic ecological opportunity created by gradual uplifts of the Andes over the last ~25 My.

ABGD: automated barcode gap discovery barcode gap= a gap between intraspecific diversity and interspecific diversity in the distribution of pairwise differences between all sequences of a typical barcode data set

ABGD: automated barcode gap discovery distlimit = a limit under which distances are statistically more likely to be intraspecific.

ABGD: automated barcode gap discovery The data set is partitioned into the maximum number of groups (i. e. species) such that the distance between two sequences taken from distinct groups will always be larger than a given threshold distance (i. e. barcode gap)

CBC: compensatory base change CBC are mutations that occur in both nucleotides of a paired structural position while retaining the paired nucleotide bond. Hemi-CBC (h. CBC) is a mutation of a single nucleotide in a paired structural position while maintaining the nucleotide bond.

CBC: compensatory base change ITS 2 exhibits a common core of RNA secondary structure throughout the Eukaryota: four helices, the third is the longest.
 CBC: compensatory")
Conserved ITS 2 secondary structure CBC species concept (multicopy, intragenomic variability !!!) CBC: compensatory base change ITS 2 sequence structure phylogenetics including RNA secondary structures improvement in the accuracy and robustness in reconstruction of phylogenetic trees CBCs never used to distinguish morphologically indistinct species CBC can correlate with species concept/diversity

- Slides: 40