General pharmacology Pharmacokinetics drug administration and absorption distribution
 Dr. Ishfaq Bukhari")
General pharmacology (Pharmacokinetics; drug administration and absorption, distribution and biovailbility Revision) Dr. Ishfaq Bukhari Pharmacology Department
, and legein (to speak or discuss)")
What is Pharmacology? From the Greek pharmakon (drug), and legein (to speak or discuss) n Broadly defined as the study of how chemical agents affect living processes. n e. g Hormones, Neurotransmitters and drugs n

Some Pharmacology Definitions and Areas of Study n n n Pharmacokinetics Study the fate of drugs once ingested. It covers , How the body absorbs, distributes, metabolizes, and excretes drugs (what the body does to a drug? ) Pharmacodynamics Study the mechanisms by which drugs work , Also study endogenous agents (what the drug does to the body? )
 by Howland Mycek n Basic and Clinical Pharmacology")
Recommended books Lippincott’s illustrated reviews (Pharmacology) by Howland Mycek n Basic and Clinical Pharmacology by by Katzung n

Routes of drug administration

Routes of drug administration

Advantages Disadvantages - Easy - Slow effect Self use -Safe - convenient - cheap - No need for sterilization - - No complete absorption - Destruction by p. H and enzymes - GIT irritation - Food - Drug interactions -Drug interactions - First pass effect - (low bioavailability). Not suitable for q vomiting & unconscious patient q emergency & bad taste drugs

Factors affecting absorption from GIT n n n n GIT motility changed by drug / diseases Presence of food Blood flow/surface area GIT juices p. H of GIT fluids Chemical/drug interactions dosage form of a drug Most of the drug is absorbed with in 1 -3 hours, mostly it occurs in small intestine , rate of absorption depends on lipid solubility , ionization and p. H.

First pass Metabolism of drug in the gut wall or portal circulation before reaching systemic circulation So the amount reaching system circulation is less than the amount absorbed Where ? Ø Liver Ø Gut wall Ø Gut Lumen Results ? Low bioavailability. Short duration of action of drugs (t ½). n
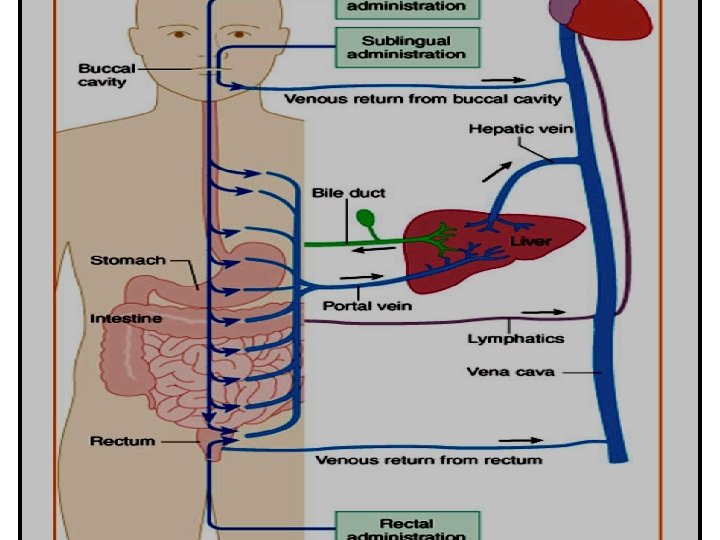

Stomach p. H n n The low p. H of the gastric contents (p. H 1– 2) may influence. e. g, the weak base diazepam will be highly Ioninized in the gastric juice, and absorption will be slow. weak acid drug, acetaminophen will exist mainly in its unionized form and can more readily diffuse from the stomach into the systemic circulation.

Small intestine n n The small intestine, with its large surface area and high blood perfusion rate, has a greater capacity for absorption than the stomach. Conditions that shorten intestinal transit time (e. g. , diarrhea) decrease intestinal drug absorption and vice versa
 n n n Tablets (enteric coated tablets) Capsules (hard")
Oral Dosage Forms (oral formulations) n n n Tablets (enteric coated tablets) Capsules (hard and soft gelatin capsules) Syrup Suspension Emulsion

Tablets Hard- gelatin capsule Spansule Soft- gelatin capsule
 q No first pass metabolism. q High bioavailability q No")
Advantages Rapid effect (Emergency) q No first pass metabolism. q High bioavailability q No GIT destruction q No food drug interaction q Dosage form: friable tablet Disadvantages Not for - irritant drugs - Frequent use

Advantages Suitable for q children q Vomiting or unconscious patients q Irritant & Bad taste drugs. q less first pass metabolism (50%) Dosage form: suppository or enema Disadvantages Irregular absorption & bioavailability. q Irritation of rectal mucosa. q
 (into skin) Subcutaneous (S. C. ) (under skin) Intramuscular (I.")
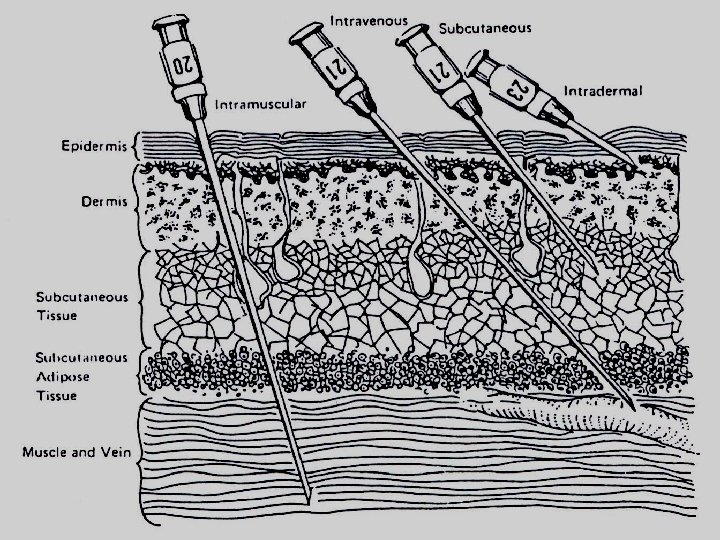
Intradermal (I. D. ) (into skin) Subcutaneous (S. C. ) (under skin) Intramuscular (I. M. ) (into muscles) Intravenous (I. V. ) (into veins) Intra-arterial (I. A. ) (into arteries) Intrathecal (I. T. ) (cerebrospinal fluids ) Intraperitoneal (I. P. ) (peritoneal cavity) Intra - articular (Synovial fluids)
 q High bioavailability q No food-drug interaction q No first")
Advantages Rapid action (emergency) q High bioavailability q No food-drug interaction q No first pass metabolism q No gastric irritation Suitable for q. Vomiting &unconsciousness q Irritant & Bad taste drugs. Dosage form: Vial or ampoule q Disadvantages Infection q Sterilization. q Pain q Needs skill q Anaphylaxis q Expensive. q Not suitable for oily solutions or poorly soluble substance
 Suitable for vaccinations")
Injection Special Utility Limitations I. D. Minute volume (0. 1 ml) Suitable for vaccinations & sensitivity test Not suitable for large volumes S. C. 0. 1 ml – 1 ml Suitable for some poorly soluble suspensions and for instillation of slow-release implants e. g. insulin zinc preparation Not suitable for large volumes I. M. larger volume 3 -5 ml Suitable Not suitable for irritant drugs for moderate volumes, oily vehicles, and some irritating substances I. V. Suitable for large volumes and for irritating substances Not suitable for oily solutions or poorly soluble substances Must inject solutions slowly as a rule

§ § § Drugs are mainly applied topically to produce local effects. They are applied to Skin (percutaneous) e. g. allergy test, topical antibacterial and steroids prep and local anesthesia Mucous membrane of respiratory tract (Inhalation) e. g. asthma Eye drops e. g. conjunctivitis Ear drops e. g. otitis externa Intranasal, e. g. decongestant nasal spray
 q")
Advantages Mucous membrane of q respiratory system q Rapid absorption (large surface area) q Provide local action q Minor systemic effect q Less side effects. q No first pass effect Disadvantages q Dosage form: volatile gases (anesthetics), aerosol, nebulizer for asthma Only few drugs can be used

Nebulizer Atomizer
")
a medicated adhesive patch applied to skin to provide systemic effect (prolonged drug action) e. g. the nicotine patches (quit smoking) Scopolamine (vestibular depressant)

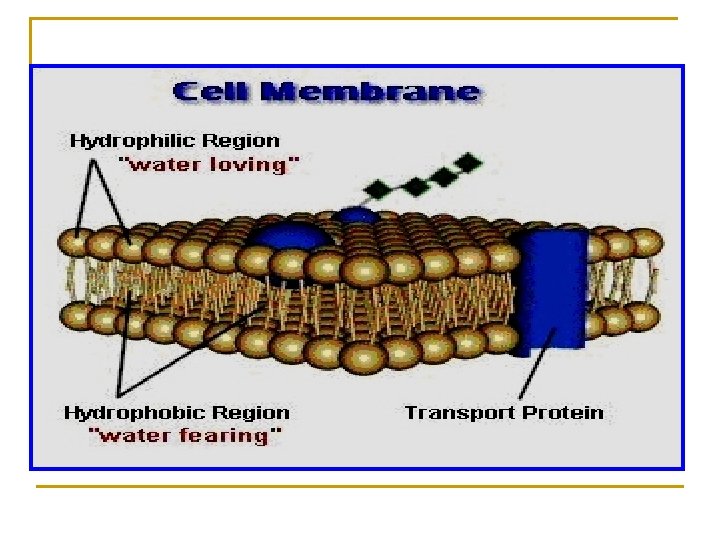
Is the passage of drug from its site of administration to its site of action through various cell membranes. n Except for intravenous administration, all routes of drug administration require that the drug be transported from the site of administration into the systemic circulation.

Is the passage of drug from its site of administration to its site of action through cell membranes. Cell membrane Sites of Administration Sites of action

n The transport of drugs across membranes occurs through one or more of the following processes: 1. 2. 3. 4. Simple diffusion = passive diffusion. Active transport. Facilitated diffusion. Pinocytosis (Endocytosis).
 is readily absorbed via diffusion through aqueous")
Ø water soluble drug (ionized or polar) is readily absorbed via diffusion through aqueous channels or pores in cell membrane. Ø Lipid soluble drug (nonionized or non polar) is readily absorbed via diffusion through lipid cell membrane itself.
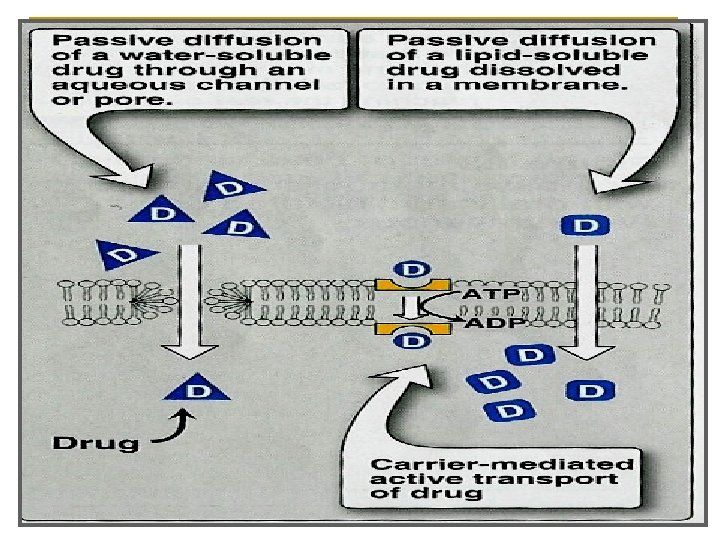
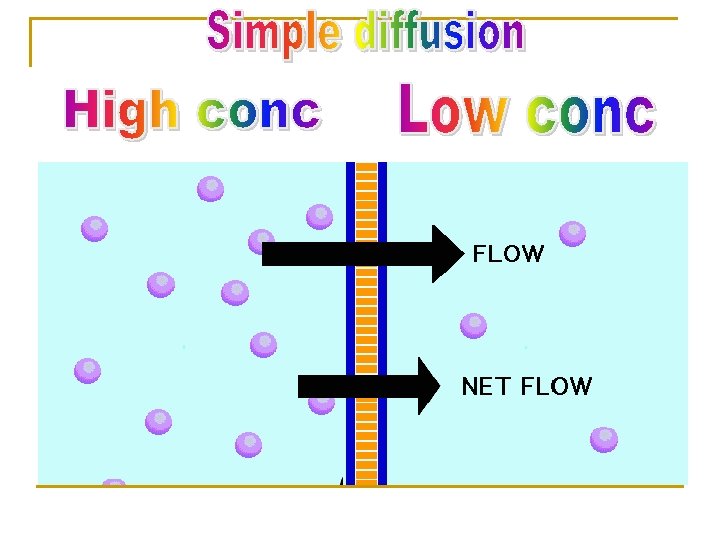

Characters Ø Ø Ø Ø common. Occurs along concentration gradient. Non selective Not saturable Requires no energy No carrier is needed Depends on lipid solubility. Depends on pka of drug - p. H of medium.
 nonionized forms (lipid soluble) in equilibrium.")
Drugs exist in two forms ionized (water soluble) nonionized forms (lipid soluble) in equilibrium. Drug ionized form + nonionized form n Only nonionized form is absorbable. n Nonionized / ionized fraction is determined by p. H and p. Ka As general basic drugs are more ionized and less diffusible in a relatively acidic medium, on the contrary basic are more lipid soluble and more diffusible in a relatively alkaline medium
: p. H at which half of")
PKa of the drug (Dissociation or ionization constant): p. H at which half of the substance is ionized & half is unionized. n The lower the p. Ka value (p. Ka < 6) of the acidic drug the stronger the acid e. g aspirin (Pka= 3. 0 ), n The higher the p. Ka value (p. Ka >8) of a basic drug, the stronger the base e. g propranolol ( p. Ka= 9. 4)
: p. H at which half of")
PKa of the drug (Dissociation or ionization constant): p. H at which half of the substance is ionized & half is unionized. p. H of the medium Affects ionization of drugs. q q Weak acids best absorbed in stomach. Weak bases best absorbed in intestine.
?")
Which one of the following drugs will be best absorbed in stomach (p. H=3)? Aspirin pka=3. 0 warfarin pka=5. 0 Arrange the following drugs in ascending order from least to greatest in rate of absorption in small intestine (p. H=7. 8)? Propranolol pka= 9. 4 Aspirin pka=3. 0

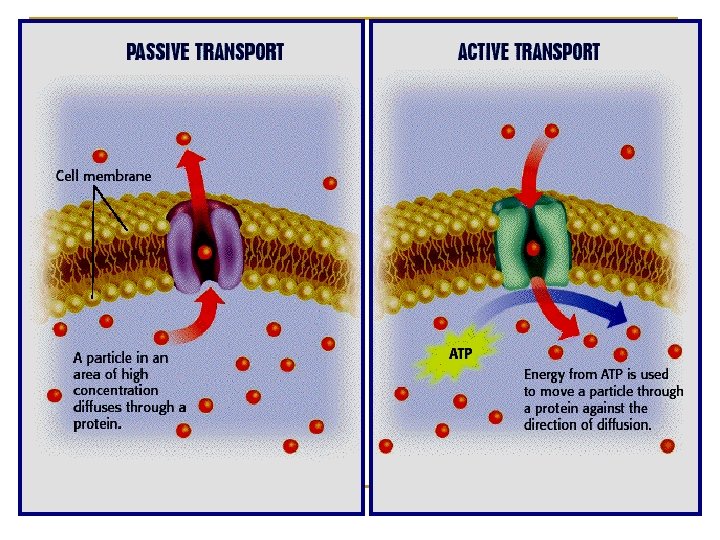
Ø Ø Ø Ø Relatively unusual. Occurs against concentration gradient. Requires carrier and energy. Specific/selective Saturable. eg. Sugar, amino acids and Iron absorption. Uptake of levodopa by brain.

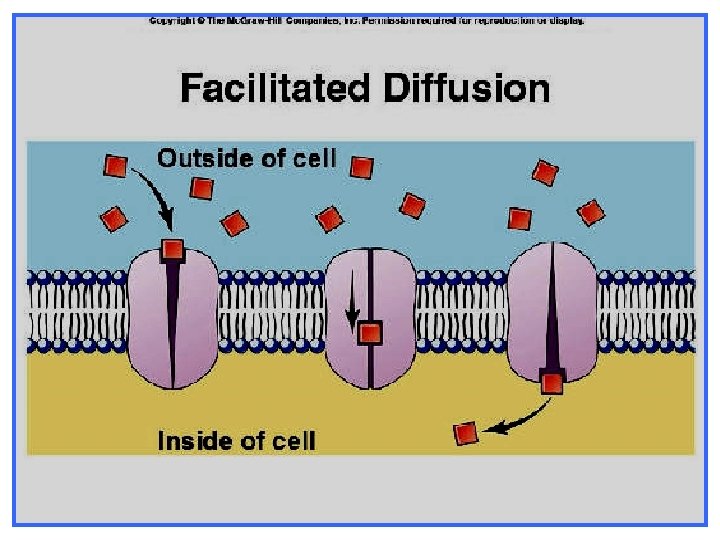
Ø Ø Ø Occurs along concentration gradient. Requires carriers Selective. Saturable. No energy is required.
 Active transport against concentration gradient")
Passive transport along concentration gradient (From high to low) Active transport against concentration gradient (From low to high) No carriers Needs carriers Not saturable Not selective Selective No energy is required
 Needs carriers Carrier-mediated facilitated diffusion")
Active transport Against concentration gradient (From low to high) Needs carriers Carrier-mediated facilitated diffusion along concentration gradient (From high to low) Needs carriers saturable Selective Energy is required No energy is required

Factors modifying drug absorption n n GENERAL FACTORS lipid solubility Degree of ionization Drug solubility (aqueous sol better than oily, susp, sol) Dosage forms (depending on particle size and disintegration) n n Concentration of drugs Circulation at site of absorption Area of absorbing surface (small intestine has large surface area) Route of administration.

Is the fraction of unchanged drug that enters systemic circulation after administration and becomes available to produce an action n n I. V. provides 100% bioavailability. n Oral usually has less than I. V.

By the end of the lectures, students should be able to define the following: Major body fluid compartments Concept of compartments. Apparent volume of distribution (vd). Plasma protein binding. Tissue binding.

Is the fraction of unchanged drug that enters systemic circulation after administration and becomes available to produce an action (therapeutic effect) n n Bioavailability (F) = AUC (oral) AUC (I. V. ) X 100

I. V. provides 100% bioavailability i. e. F= 1. n Subcutaneous, intramuscular, oral, rectal, and other extra vascular routes of administration require that the drug be absorbed first, which can reduce bioavailability. n
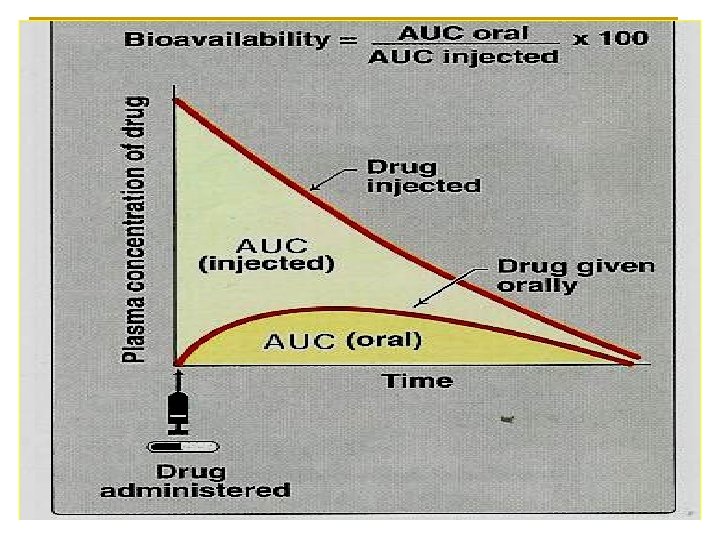

Absolute bioavailability n The bioavailability of a drug after administration by any route is compared to its intravenous standard formulation.

Relative bioavailability n is determined when two products are compared to each other, not to an intravenous standard. n This is commonly calculated in the drug industry to determine that the generic formulation is bioequivalent to another formulation. e. g Tylenol (paracetamol 500 mg) compared to panadol (paracetamol 500 mg). n

Relative bioavailability is important to get an idea of how different formulations or routes of administration differ in their bioavailability. n dosage adjustment is required when changing formulations or routes of administration. n

Bioequivalence Two drug products are considered to n be bioequivalent when the rates and extents of bioavailability of the two products are not significantly different under suitable test conditions. n Rate and extent means the amount of drugs and the time required reaching the systemic circulation.

Factors affecting bioavailability: § are the same factors controlling drug absorption GENERAL FACTORS lipid solubility Degree of ionization Drug solubility (aqueous sol better than oily, susp, sol) Dosage forms (depending on particle size and disintegration) Concentration of drugs Circulation at site of absorption Area of absorbing surface (small intestine has large surface area) Route of administration.


Is the process by which drugs leave blood circulation and enters the interstitium and/or the cells of the tissues.

Sites of Administration Absorption & distribution Elimination
 Plasma (4 L)")
Total body fluids (70% of body weight in 70 -kg individual) Plasma (4 L) Total body Fluids (42 Liters) Interstitial fluids (10 L) Intracellular volume ( 28 L)
 is the ratio of drug amount in the body")
Apparent Volume of Distribution (Vd) is the ratio of drug amount in the body to the concentration of drug in blood Vd (L)= Dose of the drug (mg) concentration in blood (mg/L) Large Vd = means long duration of action

FACTORS AFFECTING DISTRIBUTION 1. Cardiac output and blood flow. 2. Physiochemical properties of the drug. q Molecular weight q Pka. q Lipid solubility. 3. Capillary Permeability 4. Plasma protein binding 5. Tissue binding.

Blood flow to organs The greater the blood flow to tissues, the more distribution that occurs from plasma to interstitial fluids. Drugs distribute more rapidly to brain, liver and kidney > more than skeletal muscles & fat.

Physiochemical properties n Most lipid soluble drugs cross biological membranes n Hydrophilic drugs do not readily cross membranes but go through slit junctions
 Drugs with high Vd n Have higher concentrations in tissues")
Volume of Distribution (Vd) Drugs with high Vd n Have higher concentrations in tissues than in plasma. n Low molcular weight n Relatively lipid soluble. n Distributed intracellularly n Not efficiently removed by haemodialysis. n e. g. phenytion, morphine, digoxin
 Drugs with low Vd n confined to plasma & interstitial")
Volume of Distribution (Vd) Drugs with low Vd n confined to plasma & interstitial fluid. n distributed in extracellular compartments. n Polar comp or lipid insoluble drugs. e. g. Carbenicillin, vecuronium, gentamycin. n High MW e. g. heparin – insulin. n High plasma protein binding e. g. warfarin. n Do not cross BBB or placental barriers.
: Only lipid soluble drugs or actively transported drugs can cross")
Blood brain barrier (BBB): Only lipid soluble drugs or actively transported drugs can cross BBB. Hydrophilic drugs (ionized or polar drugs) can not cross BBB. Inflammation as in meningitis increase permeability to hydrophilic drugs e. g. penicillin & gentamycin Placental barrier Lipid soluble drugs can cross placental barrier and enter the fetal blood.
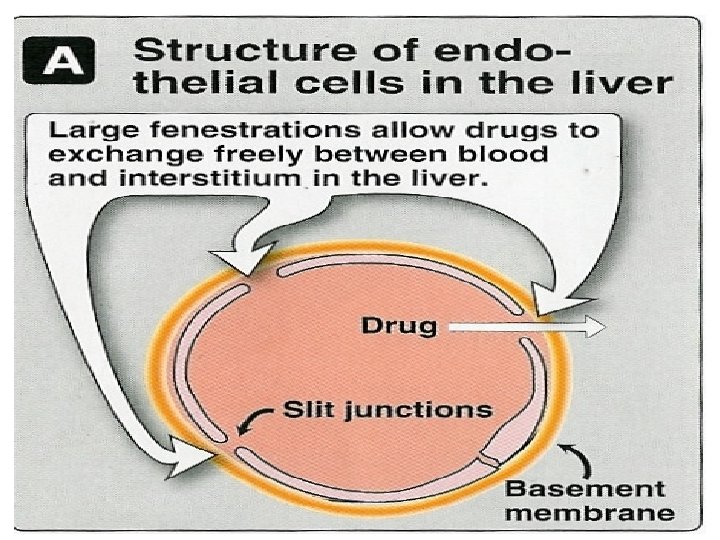
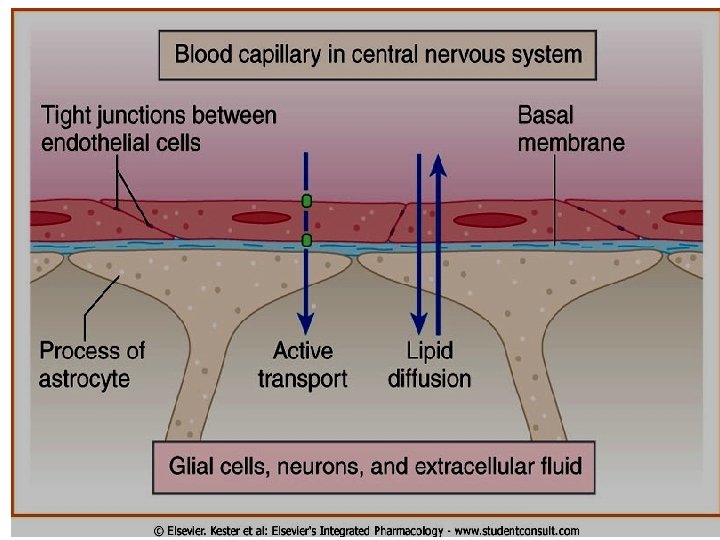

Binding of Drugs q Plasma proteins binding. q Tissue proteins binding.

Plasma Proteins Albumin Has affinity for acidic drugs as warfarin, phenytoin, aspirin

Plasma protein binding Drugs can bind to plasma proteins (acidic drug bind to albumin while basic drugs bind to glycoprotein) n Drugs exist in two forms bound and unbound forms in equilibrium n Unbound drug (free) bound drug

Tissues Binding Drugs can bind to specific tissue Tetracycline bind to bone Iodides accumulate in salivary & thyroid glands

Bound form of drug Unbound form of drug diffusible form § can not combine with § combine with receptors § active § inactive §available for metabolism § not available for metabolism & excretion § non diffusible form § § has long duration of action (t ½). §has short duration of action (t ½).

Binding of drugs and its effect on drug action n n Usually reversible. determines volume of distribution (vd) Slows drug metabolism & excretion. Prolongs duration of drug action (t 1/2). Result in clinically important drug interactions.

PHARMACOKINETICS: Pharmacokinetics: It is the study of what the body does to the drug i. e ADME n ABSORPTION n DISTRIBUTION n METABOLISM n EXCRETION of the drug Note: Pharmacodynamic is what the drug does to our body n
- Slides: 74