GENE CLONING TOOLS Genetic engineering Gene Cloning allows









 Alkaline lysis")














 sites in DNA – Restriction")








 DNA sequencing DNA mutagenesis")


- Slides: 38
GENE CLONING TOOLS
Genetic engineering Gene Cloning allows the separation and identification of a specific section of genetic material (DNA or RNA) from other sequences. It then allows the isolation of large numbers of copies of this sequence for molecular characterisation Other terms that you will see that mean the same thing include: DNA cloning molecular cloning recombinant DNA technology
What is a gene and what is a coding region? A gene is a nucleic acid sequence that code for a polypeptide or chain that has a function in an organism A gene sequence includes regulatory regions that are responsible for controlling the spatial and temporal expression of the gene product (a protein or RNA) A protein is encoded by a coding region which is the part of the gene between the translation initiation codon (normally ATG)and the translation termination codon (TAA, TGA or TAG) It is important that you appreciate the difference between a gene and a coding region. In many genetic engineering experiments we will wish to express a protein and so will only be interested in the coding region, not in the remainder of the gene from which it is derived.
Some uses of genetic engineering Cloning allows full characterisation of a gene including identification and analysis of regulatory sequences and mechanisms controlling spatial and temporal gene expression (i. e. when and where the gene is expressed) by; DNA sequence analysis Determination of 5' and 3' ends of the m. RNA transcript Location of introns/exons, Analysis of mutated forms of DNA Transcription control elements Trans-factors Sequences that control transcript stability Localisation of expressed protein Reverse genetics
Some uses of genetic engineering 2. Genome mapping and evolutionary studies 3. Expression of recombinant proteins, from the coding region, for structural and functional studies or large scale production of industrial or medical proteins 3. Protein engineering and directed evolution to generate new functional proteins 4. Diagnosis of human genetic diseases/ Forensic analysis 5. Gene therapy 6. Transgenic plants and animals
Tools and techniques We use a range of enzymes as basic tools to manipulate DNA and RNA during gene cloning and analysis processes Restriction enzymes (site-specific cutting) Phosphatases (removing 5’ phosphates) Kinases (adding 5’ phosphates) Ligases (joining fragments) Nucleases (removing DNA) Oligonucleotides (synthetic DNA eg primers and probes) DNA polymerases (replicating, amplifying) sequencing, PCR, mutagenesis eg. DNA
Tools and techniques We use various approaches to investigate genes, gene expression and to characterise where and when a gene is expressed, and where and when its product is localised and active. Gene Cloning: Vectors, enzymes, PCR, agarose gels Genes and polymorphisms: Southern blot, DNA sequencing, Next generation sequencing Transcript analysis: Northern blot, intron/exon, start site mapping, in situ hybridisation Global analysis: microarrays, proteomics, transgenic knockout/in, Next generation sequencing Protein expression profiles: western blot, immunocytochemistry, GFP, fusion proteins Protein expression studies: over-expression, functional analysis in cells Molecular interactions: immunoprecipitation, phage display, yeast hybrid systems, FRET, SPR,
Experimental design sub-cloning Non-coding regions Catalase coding region ? How can I sub-clone this catalase coding region into an protein expression vector so that I can express and purify the catalase? What steps would I need to follow and what tools/techniques would I need to use? Let’s think about what tools are available.
Key molecular biology tools 1. 2. 3. 4. 5. 6. 7. 8. Vectors Agarose gels Restriction enzymes: cut DNA Modifying enzymes: remove or add chemical group (eg phosphate or nucleotide) Ligases: join DNA Polymerases: synthesise DNA (& RNA) and/or remove nucleotides Synthetic DNA – oligonucleotides, synthetic genes Polymerase chain reaction PCR Now let’s consider a basic gene cloning flow diagram
Basic Steps in Cloning Purify vector DNA (e. g. plasmid or phage) Alkaline lysis Purify target DNA to be cloned eg genomic, c. DNA, or in silico sourced clone Digest the circular plasmid DNA with Restriction enzyme(s) Alakaline phosphatase treat the plasmid DNA to remove 5’ P’s Screen colonies to identify those with recombinant plasmid Colony PCR or plasmid isolation & restriction digest Digest the target DNA to be cloned with PCR amplify a DNA Restriction enzyme(s) fragment with carefully designed primers & digest Ligate (join) digested vector and target DNA Mixture of vector & recombinants Colonies form on plates by cell growth & plasmid replication to give a clone Transform ligation mix into competent E. coli. One cell takes up one DNA molecule Plate onto agar with antibiotic Only plasmid containing cells grow
First I need to prepare DNA for cloning PLASMID DNA c. DNA /oligo d. T purification of m. RNA http: //www. acgtinc. com/ GENOMIC DNA http: //www. pharmatech. co. kr/
First you need to prepare DNA for cloning c. DNA /oligo d. T purification of m. RNA http: //www. acgtinc. com/ We are going to recover the catalase coding region from c. DNA that is synthesised from m. RNA. The m. RNA must be isolated from the correct cells and purified (ca. 3 -5%) from other RNAs This is done by using an oligo d. T column or oligo d. T magnetic beads to isolated m. RNA which is polyadenylated. c. DNA synthesis then relies upon the enzyme Reverse transcriptase and a primer, usually an oligo d. T primer for first strand synthesis and then a self-priming or specific primer plus a DNA polymerase for second strand synthesis. If we know the gene sequences we can actually design two primers that are specific for the coding region for use in first strand c. DNA synthesis followed by PCR
Let’s assume that we are starting with a collection of oligod. Tprimed c. DNA molecules; some of these will be ones that contain our catalase sequence Catalase coding region We know the sequence of the gene from genome sequencing projects and can access this information from databases such as Genbank So we can design primers that can be used for PCR amplification of only the coding region of the c. DNA
Polymerase Chain Reaction http: //www. youtube. com/watch? v=e. Ecy 9 k_Ks. DI
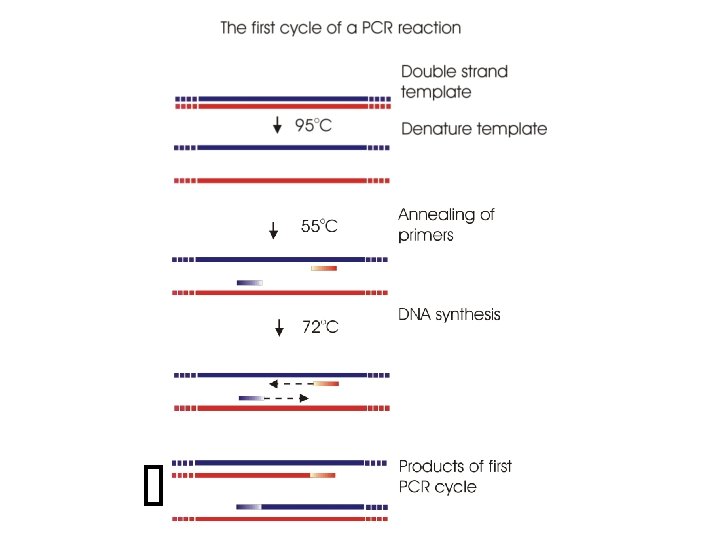
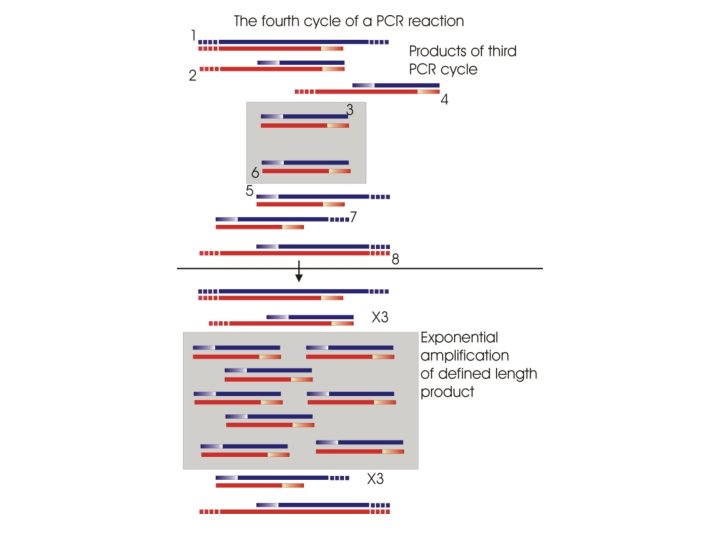
PCR involves thermal cycling – 25 -40 cycles Initial Denaturation Cycle 1 Cycle 2 94 o. C Te m p e r a t u r e Thermostable DNA polymerases: Denaturation etc DNA synthesis at high temperatures in PCR and other reactions Taq • 5’ to 3’ exonuclease and 5’ to 3’ DNA synthesis 72 o. C o 55 C Annealing Extension Time Kod, Pfu • 5’ to 3’ DNA synthesis and 3’ to 5’ exonuclease (proofreading)
Things to consider in PCR primer design • About 20 nt long primers • ~50 % GC if possible and with similar TM >55 o. C • Avoid complementary primer sequences • Avoid polypyrimidine (T, C) or polypurine (A, G) stretches • Can add sequences to 5’-end Eg. Restriction enzyme site Promoter sequence eg T 7 DNA pol can be added to the 5’ end
How do we design primers for PCR? We know the sequence of the gene from genome sequencing projects and can access this information from databases such as Genbank So we can design primers that can be used for PCR amplification of only the coding region of the c. DNA ATG 5’ TAA TGA TAG 3’ 3’ 5’ But…. how will we be able to clone this PCR amplified coding sequence into a cloning vector? First we need to decide what cloning vector we will use
Common cloning vectors Plasmids Viruses/Bacteriophage Cosmids • combination of plasmid and bacteriophage l Phagemids – combination of plasmid and bacteriophage M 13
p. ET 28 plasmid vector An example of a cloning vector used routinely in my lab Multiple cloning site Purification of expressed protein Antibiotic resistance kanamycin resistance Promoter Expressed gene regulation Origin of replication
Designing the primers for PCR? If we are going to clone into p. ET 28 we will need to add restriction sites at the ends of the coding region We do this by adding the restriction enzyme cleavage sequences to the 5’ end of the primers. If we add different sites to the 5’ and 3’ end of the coding region then we can do directional cloning so that we know the sequence is inserted into the vector in the correct orientation. We will add an Nco. I site at the 5’ end of coding region and Eco. RI site at the 3’ end 5’ 3’ Nco. I 3’ Eco. RI 5’ When we PCR amplify the coding region using these primers we will generate this sequence Nco. I Eco. RI We need to check whether we have the correct PCR product and digested vector
A more detailed look at how to design the primers for PCR of the coding region Nc o. I Ec o. R I Always label 5’ and 3’ ENDS when writing 5’ 3’ CATCTGCTAGTCCAACCTACATCATGTCGTCAAGTCAT-1 kb-ATTATTATCTCTCTGGATGTCAACATGAAACACCTGCTAACACTC 3’ GTAGACGATCAGGTTGGATGTAGTACAGCAGTTCAGTA-1 kb-TAATAATAGAGAGACCTACAGTTGTACTTTGTGGACGATTGTGAG 5’ Select primer sites (ca. 20 nt) 5’ 3’ CATCTGCTAGTCCAACCTACATCATGTCGTCAAGTCAT-1 kb-ATTATTATCTCTCTGGATGTCAACATGACACACCTGCTAACACTC GTAGACGATCAGGTTGGATGTAGTACAGCAGTTCAGTA-1 kb-TAATAATAGAGAGACCTACAGTTGTACTGTGTGGACGATTGTGAG 3’ 5’
Add sequences onto primer with a few extra 5’ nucleotides to ensure efficient restriction enzyme cleavage. Nco. I = CCATGG; Eco. RI = GAATTC 5’ 3’ CATCTGCTAGTCCAACCTACATCATGTCGTCAAGTCAT-1 kb-ATTATTATCTCTCTGGATGTCAACATGAAACACCTGCTAACACTC GTAGACGATCAGGTTGGATGTAGTACAGCAGTTCAGTA-1 kb-TAATAATAGAGAGACCTACAGTTGTACTTTGTGGACGATTGTGAG 5’ ATCC CCATGG GTCCAACCTACATCATGTCG 3’ 4 nts 3’ GACCTACAGTTGTACTTTGT GAATTCTGCT 5’ Eco. RI Nco. I Always write primers as 5’ to 3’ sequences so the reverse strand needs rewritten 5’ TCGTCTTAAGTGTTTCATGTTGACATCCAG 3’ PCR Nco. I 4 nts Eco. RI 3’ 5’
Analysing DNA fragments by agarose gel electrophoresis • Electrophoresis through an agarose gel matrix • At neutral p. H DNA and RNA have a net NEGATIVE charge due to phosphate groups and so move towards the ANODE (+ve electrode) • Small molecules move through faster than longer/larger molecules so separation is on the basis of size – For linear fragments rate of migration proportional to log 10 molecular size Can also separate on basis of conformation Plasmid DNA: Supercoiled, open circular and linear are all the same molecular size but migrate differently
Next we need to restriction digest the vector and PCR product with Nco. I and Eco. RI I o. R c E Nc o. I
Restriction enzymes Endonucleases: Digest DNA at internal (often palindromic) sites in DNA – Restriction enzymes cleave DNA only at specific recognition sites – generating fragments for cloning – map genes and polymorphisms (SNP’s) 5’ 3’ GAATTC CTTAAG 3’ CTTAA 5’ 5’AATTC 3’G 5’ 5’ 3’ 5’
Animation: Restriction enzymes • http: //highered. mcgrawhill. com/olcweb/cgi/pluginpop. cgi? it=swf: : 535: : /sites/ dl/free/0072437316/120078/bio 37. swf: : Restriction Endonucleases
Restriction enzyme sites • Restriction enzymes can leave three different types of ends – 5’ overhang (sticky end) – 3’ overhang (sticky end) – blunt Eco RI GAATTC CTTAAG G 3’ CTTAA 5’ 5’AATTC 5’ OVERHANG 3’G Pvu II Kpn I CAGCTG GTCGAC GGTACC CCATGG CAG 3’ GTC 5’ 5’CTG 3’GAC BLUNT END GGTAC 3’ C 5’ 5’C 3’CATGG 3’ OVERHANG The ends generated allow different DNA fragments to be joined
Restriction enzyme sites • Some enzymes recognise different sites but generate the SAME sticky ends Bam HI Bgl II GGATCC CCTAGG AGATCT TCTAGA Bam HI end G 3’ CCTAG 5’ Bgl II end 5’GATCT + 3’A Sau 3 A NGATCN NCTAGN Product GGATCT CCTAGA Will not cut with Bam HI or Bgl II, but will still cut with Sau 3 A
Often the restriction digested vector DNA is also treated with the enzyme • Alkaline phosphatase: – removes the 5’ phosphate groups from DNA, normally the vector DNA – needs inactivated usually by heat before the ligation step (otherwise it can dephosphorylate the insert as well!!) Hydrolysis of phosphate ester -3 + 2 PO 4 Gene Cloning and DNA Analysis by T. A. Brown. © 2006 T. A. Brown.
Why do we use alkaline phosphatase? Vector plus insert 3’OH 5’PGATC CATG 5’ 3’OH P GATC CATG Vector with NO insert Insert Ligation 3’OH 5’OH CATG 5’OH GATC Vector 3’OH GATC CATG No ligation
The vector and insert DNA are then mixed in a ligase buffer containing ATP and DNA ligase • Generates phosphodiester bonds between 3’OH and 5’ P • Must have a 5’Phosphate (5’P) for ligase to function Join two double strand molecules together if they have suitable ends Vector 3’OH 5’PGATC CATGP 5’ 3’OH GATC CATG Repair single-strand breaks in the phosphodiester backbone useful in some site-directed mutagenesis applications 3’OH 5’P O-P-O Insert
Following a ligation reaction an aliquot is transformed into competent E. coli cells. The E. coli cells are treated with Ca. Cl 2 or Rb. Cl 2 to disrupt their cell walls and can be stored frozen at -80 o. C. For a transformation reaction aliquots of cells are thawed on ice and DNA is added, typically around 40 -5 0 ng. After incubating on ice the cells are heat shocked for around 1 -2 min at 42 o. C so that cells take up the DNA Very few of the cells will actually become transformed and so we need to be able to identify those cells that have been transformed and we do this by antibiotic selection
Selection and screening All the transformed colonies will contain a vactor, but NOT all will contain recombinant plasmids (1) Clones containing vector molecules can grow – they are antibiotic resistant! • Reduce vector only (eg alkaline phosphatase) (2) How do you identify recombinants? Blue white selection (based on lac. Z activity) Colony PCR Purify plasmid & restriction digest Hybridization screening Most common then DNA sequence
DNA Polymerases Uses: DNA synthesis (and sometimes as an exonuclease) DNA sequencing DNA mutagenesis DNA labelling E. coli DNA polymerase I: synthesises DNA using a template and primer – Three activities: • • • 5’-3’ exonuclease (repair) 5’-3’ DNA synthesis 3’-5’ exonuclease (proof-reading) Klenow fragment T 4 DNA pol T 7 DNA pol These are useful for some DNA manipulation including • Filling in sticky ends to make them blunt ends • Radioactive labelling ends • DNA synthesis reactions that use a primer
DNA Polymerases that are now more commonly used than Pol I • T 7 and T 4 phage DNA polymerases: – • Klenow activities, but more efficient Thermostable DNA polymerases: – DNA synthesis at high temperatures in PCR and other reactions Taq – • – Kod, Pfu • • 5’ to 3’ flap exonuclease and 5’ to 3’ DNA synthesis and 3’ to 5’ exonuclease (proofreading) Reverse transcriptase: – synthesises c. DNA using RNA as a template and a DNA primer
Reading associated with this lecture 1. Gene cloning essentials 3 1. 1 Introduction 4 1. 2 Gene cloning applications 4 1. 3 Gene cloning in the laboratory 5 1. 4 Gene cloning processes 14 1. 5 Further types of gene cloning 18 1. 6 Chapter summary 21 2. Polymerase chain reaction 23 2. 1 Introduction 23 2. 2 How PCR works 24 2. 3 The PCR protocol 26 2. 4 PCR techniques and applications 31 2. 5 Forensic DNA analysis 40 2. 6 Future prospects 41 2. 7 Chapter summary 41