Functions of some Proteins Catalysis enzymes Structural collagen

 Structural (collagen) Contractile (muscle) Transport (hemoglobin) Storage (myoglobin)")
 – sequence of amino acids")










![Acid Ionization Constant [CH 3 COO-][H+] Ka = ---------[CH 3 COOH] [CH 3 COO-]](https://slidetodoc.com/presentation_image_h/c344e5e07000ab702291c1c8eb3ca162/image-22.jpg "Acid Ionization Constant [CH 3 COO-][H+] Ka = ---------[CH 3 COOH] [CH 3 COO-]")







 of an amino acid")



 The sequence of amino acids (N-term to C-term) in a")











 The two most common secondary structures are the a-helix and")











")



 The three dimensional folding of a polypeptide is its tertiary")





 is an assembly of 3 o structures (two or more")














- Slides: 83
Functions of some Proteins Catalysis (enzymes) Structural (collagen) Contractile (muscle) Transport (hemoglobin) Storage (myoglobin) Electron transport (cytochromes) Hormones (insulin) Growth factor (EGF) DNA binding (histones) Ribosomal proteins Toxins and venoms (cholera & melittin) Vision (opsins) Immunoglobins
Levels of Protein Structure • Primary structure (1 o) – sequence of amino acids starting from the N-terminus of the peptide. • Secondary structure (2 o) – conformations of the peptide chain from rotation about the a-Cs, e. g. a-helices and b-sheets, etc. • Tertiary structure (3 o) – three dimensional shape of the fully folded polypeptide chain. • Quaternary structure (4 o) - arrangement of two or more protein chains into multisubunit molecule
Levels of Protein Structure Bonding: 1 o = covalent 2 o = H-bond 3 o = covalent & noncovalent 4 o = noncovalent
Amino Acids The 20 common are those used in making protein on a ribosome using m. RNA and t. RNA. These are called a-amino acids since each has a carboxyl group and an amino group attached to an a-carbon atom. They differ by the sidechain or “R” group. a +NH -CH-COOH 3 l R
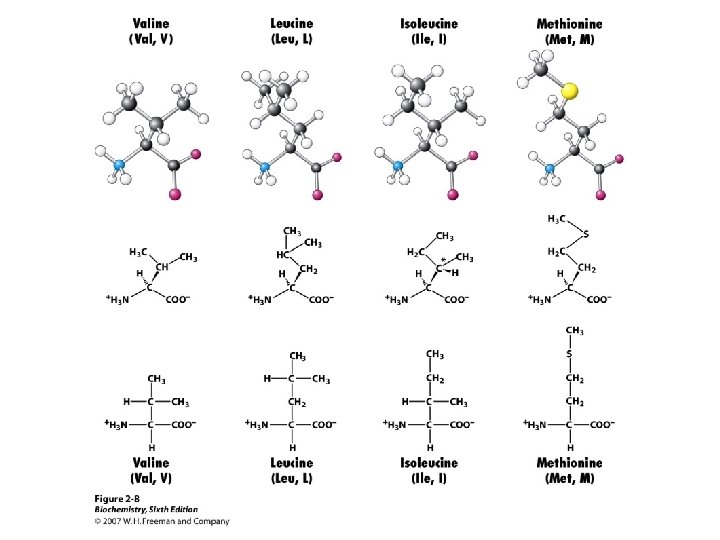
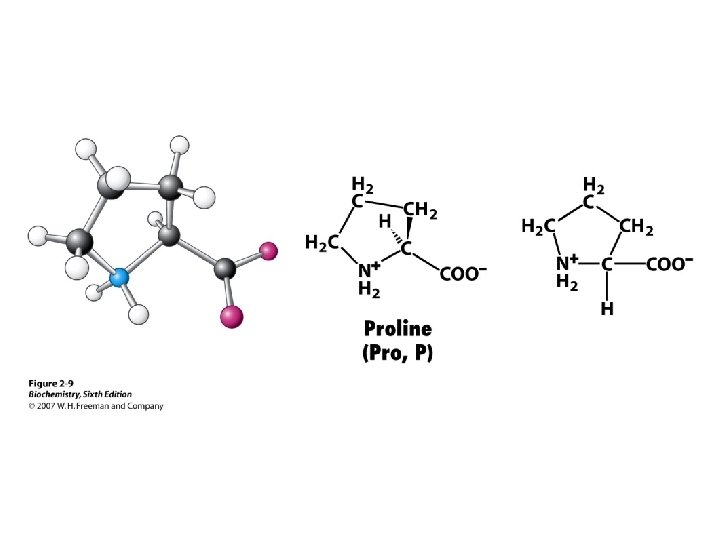
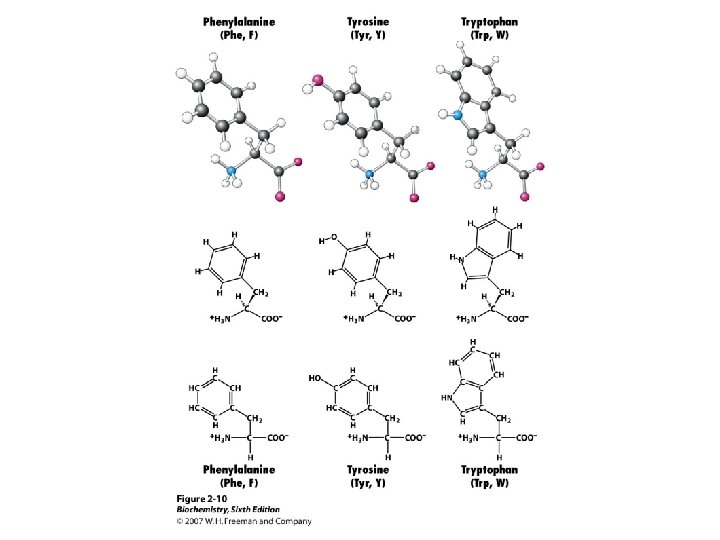
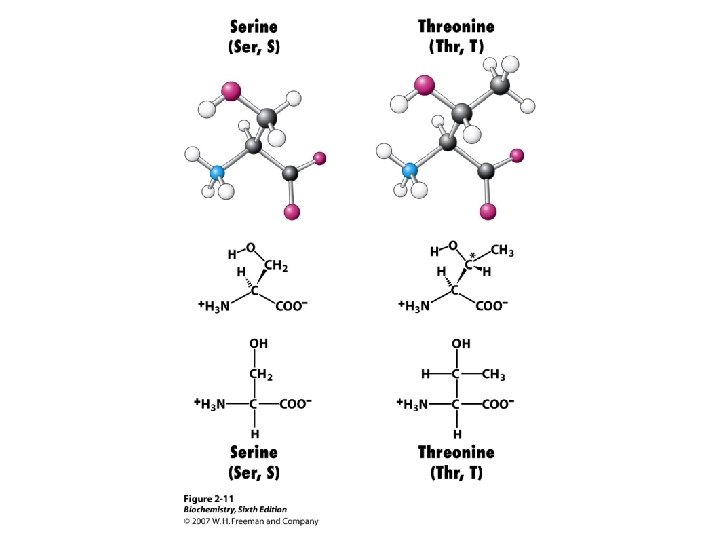
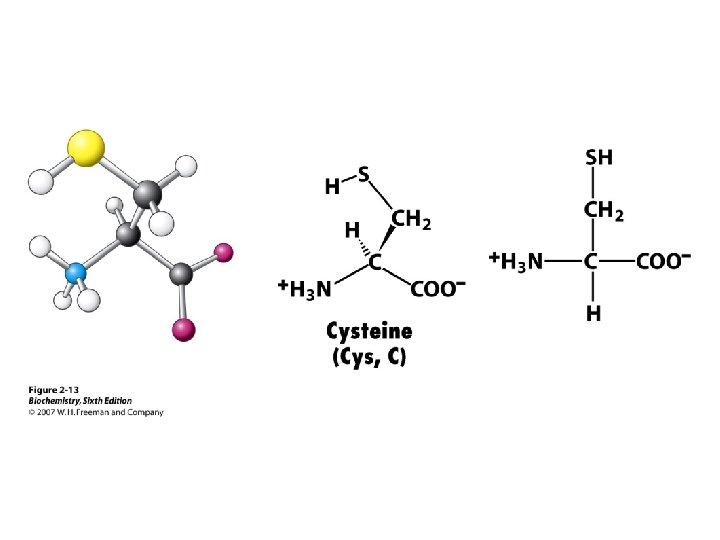
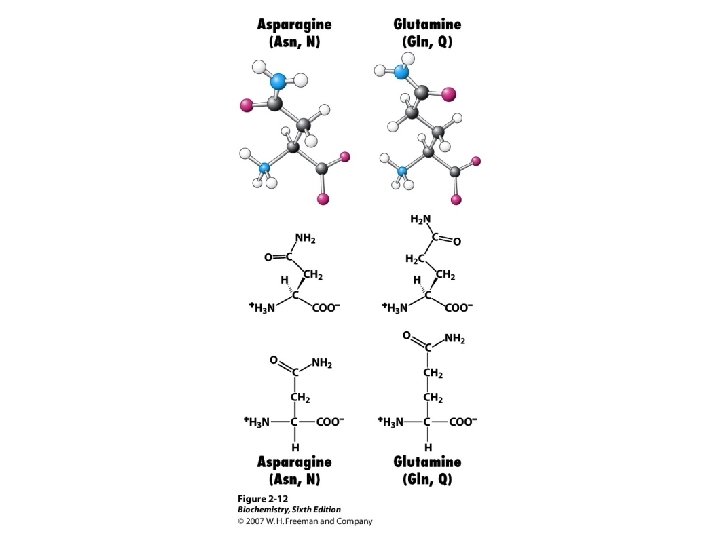
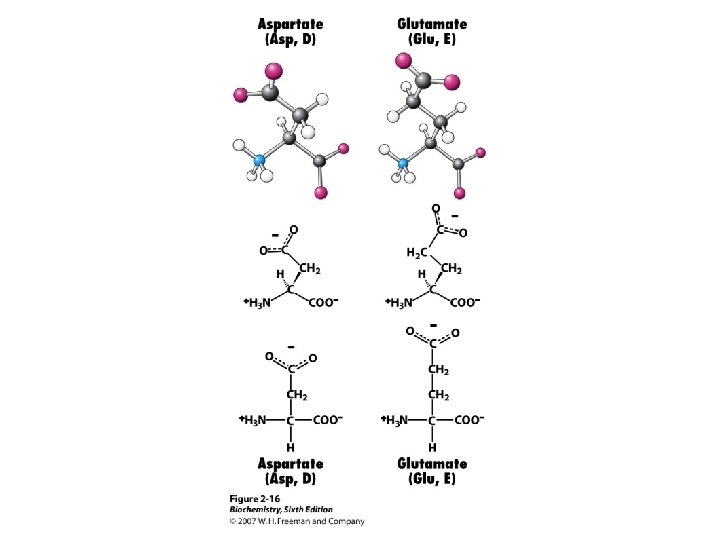
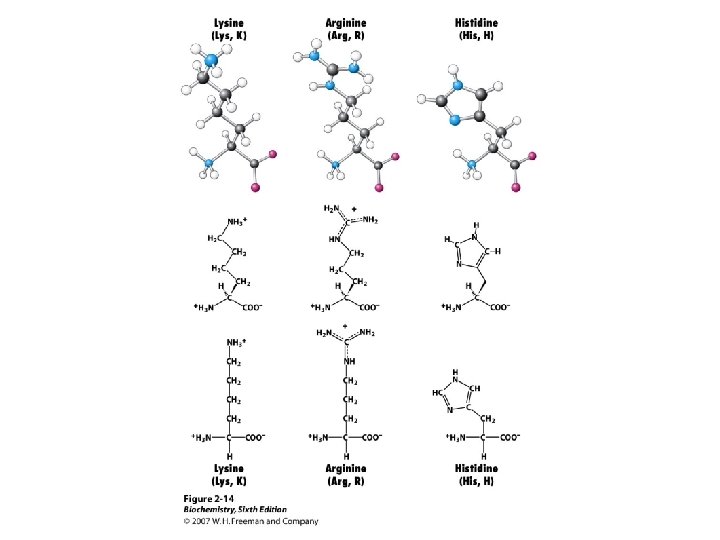
Amino Acid Classification is made using the structure of the side chain, R. (* = essential) 1. None (hydrogen): 2. Non-polar: Aliphatic: Alicyclic: 3. Aromatic: 4. Polar uncharged: 5. Thiol: 6. Acidic: 7. Basic: Gly Ala, Val*, Leu*, Ile*, Met* Pro Phe*, Tyr, Trp* Ser, Thr*, Asn, Gln Cys Asp, Glu Lys*, His*, Arg*
ball & stick model wedge Fischer projection
Amino Acid Names & Codes
All are Chiral at the a-carbon atom except Gly
D, L Assignments The convention for making D, L assignments is to draw a Fischer projection with the carboxyl at the top and the R at the bottom. +NH 3 to the left = L to the right = D
L stereochemistry = S, for this case
Acids and Bases, p. H, p. Ka • p. H is defined as the negative logarithm of the concentration of H+ p. H = - log [H+] = log 1/ [H+] • p. Ka is defined as the negative logarithm of an acid ionization constant Ka. p. Ka = - log [Ka] = log 1/ [Ka]
Acetic Acid is a Weak Acid • Weak acids and bases do not ionize (dissociate) completely in H 2 O. CH 3 COOH <==> CH 3 COO- + H+ acetic acid acetate anion conjugate acid form conjugate base form
Acid Ionization Constant [CH 3 COO-][H+] Ka = ---------[CH 3 COOH] [CH 3 COO-] -log Ka = -log [H+] + -log --------[CH 3 COOH] [CH 3 COO-] p. H = p. Ka + log --------[CH 3 COOH]
Henderson-Hasselbalch Written in a more general form this expression is called the Henderson-Hassebalch equation. This relates the p. H of a solution to the p. Ka of the weak acid in solution and the ratio of its conjugate base/conjugate acid forms. [A-] p. H = p. Ka + log -----[HA]
Buffers Resist Change in p. H • Buffer capacity is the ability of a solution to resist changes in p. H. • Most effective buffering occurs where: solution p. H = buffer p. Ka • At this point: [conjugate acid] = [conjugate base] • Effective buffering range is usually at p. H values equal to the p. Ka ± 1 p. H unit
Amino Acid Ionization, p. Kas Each of the 20 common a-amino acids has two p. Ka values, for the carboxyl group and the amino group attached to the a-carbon. +NH -CH -COO- + H+ -CH -COOH < === > 3 2 +NH - < === > NH -COO- + H+ -CH -COO 3 2 2 2 Seven of the 20 have an ionizable sidechain and therefore have a third p. Ka value.
Amino Acid p. Kas Gly Ala Val Leu Ile Met Pro Phe Trp Ser Thr Asn Gln Cys Asp Glu Tyr Lys His Arg a-COOH 2. 34 2. 32 2. 36 2. 28 1. 99 1. 83 2. 21 2. 63 2. 02 2. 17 1. 71 2. 09 2. 19 2. 20 2. 18 1. 82 2. 17 a-+NH 3 9. 60 9. 69 9. 62 9. 60 9. 21 10. 60 9. 13 9. 39 9. 15 10. 43 8. 80 9. 13 10. 78 9. 82 9. 67 9. 11 8. 95 9. 17 9. 04 R (sidechain) 8. 33 3. 86 4. 25 10. 07 10. 79 6. 00 12. 48
Peptide 3. 86 D, 4. 25 E Peptide 10. 0
Effect of Change in p. H
The Structure of an Amino Acid An amino acid can never exist as an uncharged compound. © 2014 Pearson Education, Inc.
The p. I of Alanine The isoelectric point (p. I) of an amino acid is the p. H at which it has no net charge. © 2014 Pearson Education, Inc.
The p. I of Lysine © 2014 Pearson Education, Inc.
The p. I of Glutamic Acid © 2014 Pearson Education, Inc.
Formation of a Peptide Two amino acids are joined together to form a peptide (amide) bond with a loss of HOH. After becoming part of a peptide or protein these are called “residues” due to loss of HOH.
Primary Structure (1 o) The sequence of amino acids (N-term to C-term) in a peptide or protein is its primary structure. A pentapeptide
Disulfide Bond Formation
Disulfide Bridges Disulfide bridges contribute to the overall shape of the protein. © 2014 Pearson Education, Inc.
Straight and Curly Hair © 2014 Pearson Education, Inc.
Insulin has • two interchain disulfide bridges and • one intrachain disulfide bridge. © 2014 Pearson Education, Inc.
Peptide Bond Resonance Due to resonance participation of the unshared pair of electrons on N, amides are neutral. . .
Peptide Bond Planarity 6 atoms are coplanar
Peptide Bond Structures Less crowded in the favored trans arrangement
Rotation Sites, y and f The rotational arrangements about a-carbons of a peptide or protein gives its secondary (2 o) structure.
f View from N-term to a-carbon
y View from a -carbon to C-term
Ramachandran Plot
Secondary Structure (2 o) The two most common secondary structures are the a-helix and the b-sheet. Each of these 2 o structures have fairly specific y and f angles. All other rotational angles represent “random” secondary structure. Secondary structure is maintained by hydrogen bonding. a-helix by intramolecular H-bonds b-sheet by intermolecular H-bonds
a-helix in a Ramachandran Plot y = -47 f = -57
The a-helix, a 3. 613 helix
The a-helix
Hydrogen Bond Contacts
a-helix in a Protein
b-sheet in a Ramachandran Plot y f = +135 = -139
A b-sheet strand
Anti-parallel b-sheet
Parallel b-sheet
Anti-parallel b-sheet
b-sheet in a Protein
b-turn or hairpin turn A turn is 4 -5 aa residues. A b-turn (hairpin) is 4 aa residues.
Loops are usually larger than turns. >5 aa residues
a-Keratin, a fibrous protein, forms an a coiled coil Each strand is a modified a-helix, 3. 5 residues/turn. Right-handed helices form a left-handed supercoil.
Collagen, a triple helix Gly every third residue; Gly-Pro-HPro is frequent. Interstrand H-bond at Gly. No Cys, so, no -S-S-
Tertiary Structure (3 o) The three dimensional folding of a polypeptide is its tertiary structure. Both the a-helix and b-sheet may exist within the tertiary structure. Generally the distribution of amino acid sidechains in a globular protein finds mostly nonpolar residues in the interior of the protein and polar residues on the surface. Tertiary structure is maintained by noncovalent interactions and disulfide bonding.
Myoglobin, a globular protein
Myoglobin Surface Blue = charged Cross-section Yellow = hydrophobic
Porin, a membrane spanning protein
Domain A domain is a discrete globular area within protein. There are four general types: All a All b Mixed a/b a+b only a- helices and loops only b- sheet & loops or turns alternating abab cluster of a then cluster of b
Domain Multiple domains exist in the protein below.
Quaternary Structure (4 o) is an assembly of 3 o structures (two or more subunits). A dimer
Hemoglobin, a tetramer Quaternary structure is maintained by noncovalent interactions
Denaturing Proteins Denaturing agents destroy the protein 3 o structure (causes the protein to unravel). Methods: Heat; Extremes of p. H; Detergents; Mechanical agitation; Mercaptoethanol – breaks -S-S- bonds; 6 M guanidine HCl or 10 M urea: these are chaotropic agents that break up noncovalent interactions.
Denaturing agents
Disulfide oxidation-reduction Oxidized Reduced
Ribonuclease 4 disulfide bonds. -S-S-
Then removing urea and ME permits reoxidation.
Reoxidation reforms –S-S- but not necessarily in the correct place. A trace of ME allows reduction/oxidation to occur until the low free energy form is found and >98% of activity is restored.
Sharp Transition suggests an all or nothing effect in denaturing.
Lysozyme Ribbon diagrams 3 o structure Active site & -S-S-
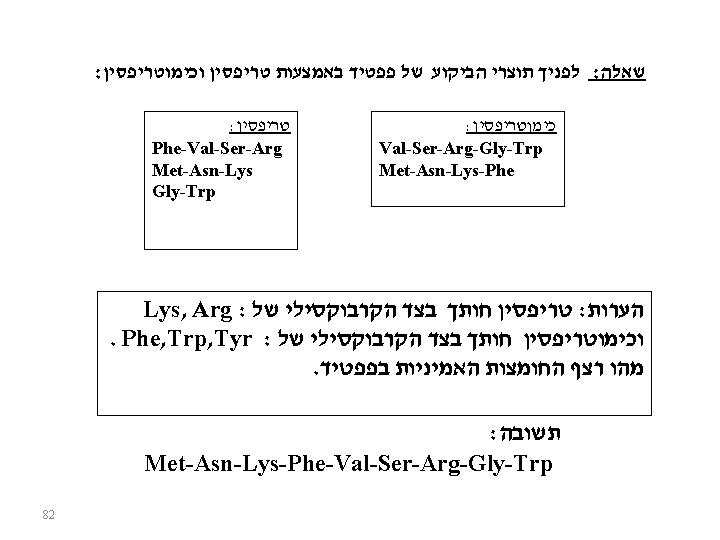
Protein Cleavage Protein sequencing is most manageable with small polypeptides. Therefore, in order to sequence a large protein, it must be cleaved into smaller pieces. Cleavage is conducted using either chemical or enzymatic methods. The pieces must be separated and purified before sequencing.
Chemical and Enzymatic Cleavage
CNBr Cleavage at Met
Enzymatic cleavage by Trypsin
An Example, Peptide overlap