Fonones y Elasticidad bajo presin ab initio Alfonso

Fonones y Elasticidad bajo presión ab initio. Alfonso Muñoz Dpto. de Física Fundamental II Universidad de La Laguna. Tenerife. MALTA Consolider Team Canary Islands, SPAIN

Plan de la charla: Ø Introducción : Ab initio methods Ø Fonones. Propiedades dinámicas. Ejemplos Ø Elasticidad estabilidad mecánica bajo presión Ø Conclusiones.

Ab initio methods Ø State of the art Ab Initio Total Energy Pseudopotential calculations are useful to study many properties of materials. Ø No experimental input required (even the structure). Only Z is required. They can provide and “predict” many properties of the material if the approximations are correct! Ø DFT is the standar theory applied, it is “exact” but one need to use approximations, XC functional (LDA, GGA etc…), BZ integration with k-special points, etc. (some problems in high correlated systems, f-electrons etc. . ). DFPT also available, allows to study phonons, elastic constants etc… Ø More elaborated approximations are also available, like LDA + U, MD, etc. . Ø Many computer programs available, some times free (Abinit, quantum espresso, VASP, CASTEP, etc…) Ø Ab initio methods provide and alternative and complimentary technique to the experiments under extreme conditions.

"Those who are enamoured of Practice without Theory are like a pilot who goes into a ship without rudder or compass and never has any certainty of where he is going. Practice should always be based upon a sound knowledge of Theory. “ Leonardo da Vinci, (1452 -1519 )

Well tested:
")
. “Prediction is very difficult, especially about the future”. Niels Bohr (1885 -1962)

Fonones y espectroscopía, ¿para que? Thermal Expansion Elasticity - deformation Superconductivity Thermal Conductivity

Lattice Dynamics Lattice Potential: Harmonic approximation: Hooke’s law! IFC

Phonons: linear chain of atoms Harmonic approximation: Ansatz: =>

Phonons: linear chain of atoms Linear chain of atoms

Linear chain with two different "spring constants" two atoms per unit cell Ansatz:
 and optic")
Phonons Linear chain with two different "spring constants" Two solutions: acoustic (-) and optic (+) branches

3 D Phonon Dispersion Relations 3 THz ~ 100 cm-1 ; 1 me. V ~ 8 cm-1 THz Si j = 3 N branches cm-1 3 C-Si. C LO TO G. Nilsson and G. Nelin, PRB 6, 3777 (1972) W. Weber, PRB 15, 4789 (1977) J. Serrano et al. , APL 80, 23 (2002)

Polar crystals: LST relation Ionic crystals: Macroscopic electric field Lyddane-Sachs-Teller relation Born effective charges X. Gonze and C. Lee, PRB 55, 10355 (1997)
")
Anisotropy: crystal field Ga. N T. Ruf et al. , PRL 86, 906 (2001)

Anisotropy: Selection Rules Not all modes are visible with the same technique! B 1: SILENT modes Not allowed modes are visible at the same time! J. M. Zhang et al. , PRB 56, 14399 (1997)
 (2006)")
Elasticity h-BN Christoffel equations A. Bosak et al. , PRB 73 041402(R) (2006)

Vibrational spectroscopies Probes: Light: photons Particles • Brillouin spectroscopy • Raman spectroscopy • Infrared absorption spect. • Inelastic X-ray Scattering Ø electrons: High Resolution e- Energy Loss Ø He: He atom scattering Ø neutrons • Time-of-flight spectroscopy • Inelastic Neutron Scattering

Vibrational spectroscopies

Vibrational spectroscopies Brillouin spec. • Excitations of 2 me. V-0. 6 me. V • Acoustic phonon branches at low q (sound waves) • Information: E Vs (sound speed) linewidth Atenuation X-ray scattering • Excitations ~ me. V, ~ Å-1 • whole BZ available àNo kinematics restrictions à Dispersion + r( ) • Energy resolution ~1 me. V Optic b. • 1 me. V-e. V Excitations • Optic phonons at the center of the Brillouin zone • High resolution • Different selection rules Acoustic branches Raman spec. Neutron scattering • Excitations ~ me. V, ~ Å-1 • whole BZ available àDispersion + r( ) Kinematical limit: vs < 3000 m/s

Infrared Spectroscopy Absorption spectroscopy: dipolar selection rules Target: polar molecular vibrations, determination of functional groups in organic compounds, polar modes in crystals

BILBAO CRYSTAL… SERVER SAM
 SMODES, FINDSYM, ETC……. .")
ISOTROPY PACKAGE (STOKES ET AL. ) SMODES, FINDSYM, ETC……. .

Interatomic force constants Born-Oppenheimer approximation In the harmonic approximation, the total energy of a crystal with small atomic position deviations is: where the matrix of IFC’s is defined as: 25

Physical Interpretation of the Interatomic Force Constants The force conjugate to the position of a nucleus, can always be written: We can thus rewrite the IFC’s in more physically descriptive fashion: The IFC’s are the rate of change of the atomic forces when we displace another atom in the crystal. 26

Dynamical properties under pressure. . The construction of the dynamical matrix at gamma point is very simple: Phonon dispersion, DOS, PDOS requires supercell calculations. Also DFPT allows to include T effects, Thermod. properties, etc…

Relation between the IFC’s and the dynamical matrix The Fourier transform of the IFC’s is directly related to the dynamical matrix, The phonon frequencies are then obtained by diagonalization of the dynamical matrix or equivalently by the solution of this eigenvalue problem: phonon displacement pattern masses square of phonon frequencies 28

Phonon band structure of α-Quartz Si. O 2 9 atoms per unit cell Nb. of phonon bands: Nb. of acoustic bands: 3 Nb. of optical bands: Polar crystal : LO non-analyticity Directionality ! [X. Gonze, J. -C. Charlier, D. C. Allan, M. P. Teter, PRB 50, 13055 (1994)] 29

LO-TO splitting Wrong behaviour High - temperature : Fluorite structure ( , one formula unit per cell ) Supercell calculation + interpolation ! Long-range dipole-dipole interaction not taken into account Calculated phonon dispersions of Zr. O 2 in the cubic structure at the equilibrium lattice constant a 0 = 5. 13 Å. [From Parlinski K. , Li Z. Q. , and Kawazoe Y. , Phys. Rev. Lett. 78, 4063 (1997)] DFPT (Linear-response) with = 5. 75 = -2. 86 and = 5. 75 LO - TO splitting 11. 99 THz Non-polar mode is OK 30

Thermodynamic properties In the harmonic approximation, the phonons can be treated as an independent boson gas. They obey the Bose-Einstein distribution: The total energy of the gas can be calculated directly using the standard formula: Energy of the harmonic oscillator Phonon DOS All thermodynamic properties can be calculated in this manner. Note: 2 April 2008 ISVS 2008: Phonon Bands and Thermodynamic Properties 31
 Tb. PO 4 , Dy.")
1. Even with f electrons (PRB 85, 024317 (2012) Tb. PO 4 , Dy. PO 4
 Cardona, …Muñoz. et al.")
Zn. S, Phys. Rev B 81 075207 (2010) Cardona, …Muñoz. et al.
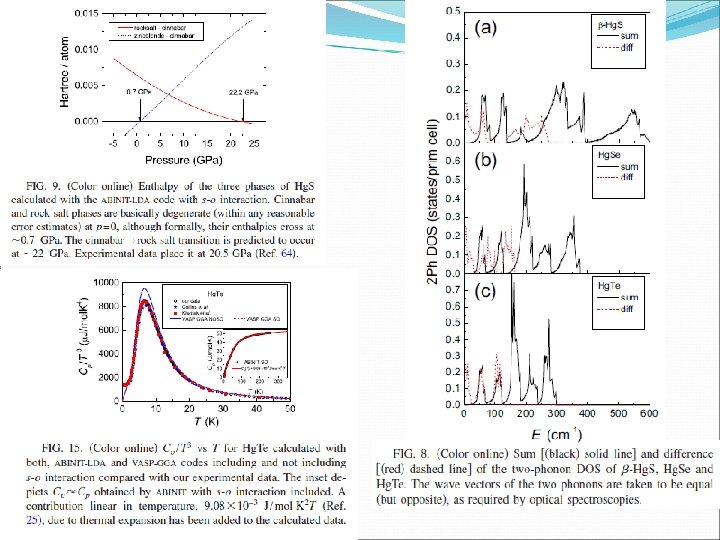

Spin-orbit, phonon dispersión, temperature effects, etc…. DFPT Inverted s-o interaction. Contribution of the negative splitting of 5 d G 15 states of Hg wich overcompesate the positive splitting of the S 3 p.

Cu. Ga. S 2 electronic and phononic properties Eficient photovoltaic materials. (Phys. Rev. B 83, 195208 (2011) ) Chalcopyrite tetragonal SG I-42 d Few it is know about this compounds. We did a structural , electronic and phononic study of thermodynamical properties Two main groups of chalcopyrites: • I-III-VI 2 derived from II-VI zb compounds (Cu. Ga. S 2, Ag. Ga. S 2. . ) • II-IV-V 2 derived from III-V zb comp. ounds (Zn. Ga. As 2, …. ) Two formula units per primitive cell We will focus on the study of some thermodynamics properties, like the specific heat with emphasis in the low-T region where appear strong desviation of the Debye T 3 law, phonons, etc…

silicon zincblende, Zn. SS chalcopyrite, Cu. Ga. S 2
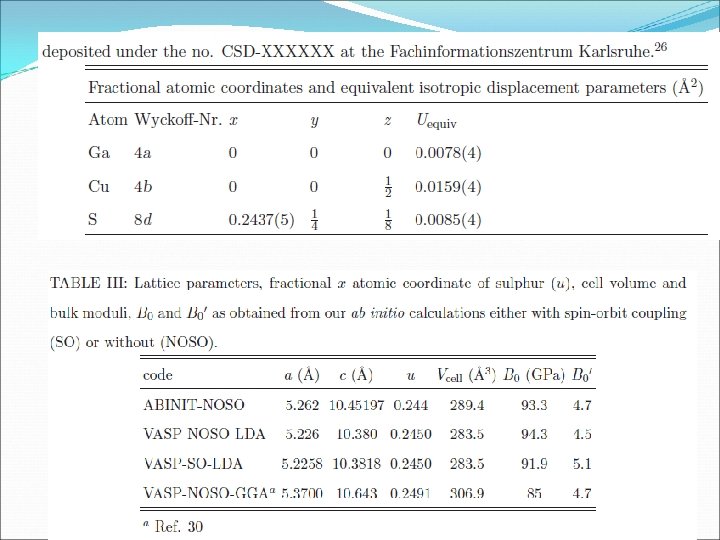
")
Elastic Constants Cij (no experimental data available)

5 Phonons Starting from the electronic structure we calculate the phonon dispersion relations with density functional perturbation theory. We compare them with Raman and IR measurements at the center of the BZ (see Figure). Cu. Ga. S 2 comparison with Raman and IR measurements ( ) shows good agreement inelastic neutron scattering data are not available as yet
 Through BZ integration of")
7 Phonon Density of States PDOS (states / formula unit) Through BZ integration of the phonon dispersion relations the phonon density of states (total or projected on the individual atoms Cu, Ga, S)) are obtained (see Figure). sulphurlike Cu. Ga Ga. Cu below 120 cm-1: essentially Cu- and Ga-like phonons above 280 cm-1: essentially S-like phonons midgap feature at ~180 cm-1: Ga-, Cu -like The partial density of states are useful for calculating the effect of isotope disorder on the phonon linewidths

Two-phonon No second-order Raman spectra available. The calculated sum an difference densities will help to interpret future measured spectra. It is posible to establish a correspondence between the calculated two-phonon Raman spectra of Cu. Ga. S 2 and other two-phonon measured spectra of binary compounds.
 for a (cubic) crystal can be expressed")
The effect of phonons the on Vo(T) for a (cubic) crystal can be expressed in terms of mode Grüneisen parameters γqj : Due to the large number of phonons bands, a first approximation is to use only the values at the Zone center for the evaluation of the termal expansion coefficient. The temperature dependence of Vo for q= 0 is: Or from thermodyn… using S(P, T)

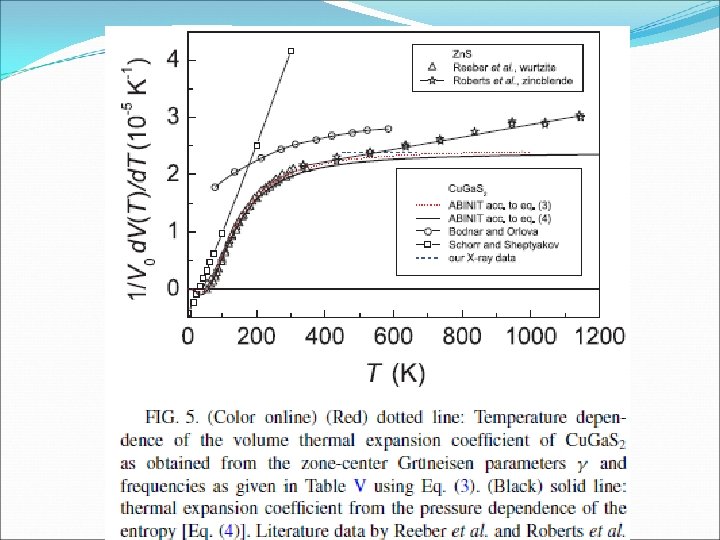
, and the")
Heat capacity The phonon DOS allows to calculate the Free Energy F(T), and the specific heat at constant volume And the constant pressure Cp can be obtained:
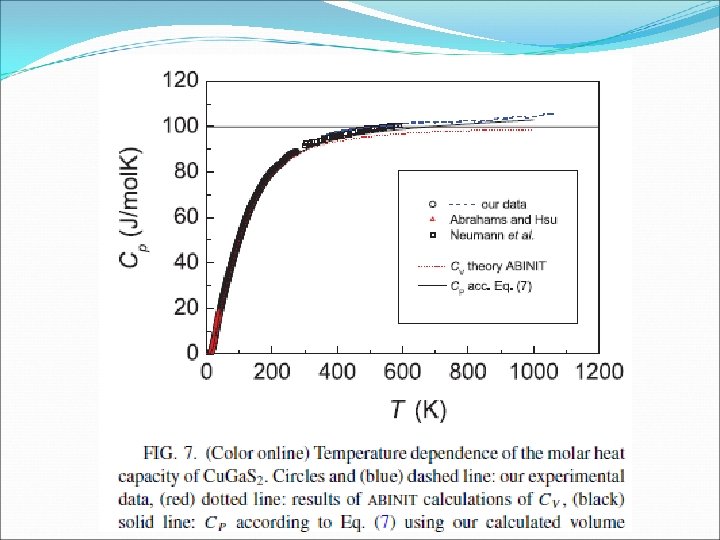

Comparison of calculated and measured specific heat Cu. Ga. S 2 versus Ag. Ga. S 2 q. Debye(0) = 355 K 8 peak at ~ 20 K in CP /T 3 representation from Cu/Ga like phonons (ratio 1: 6 to lowfrequency peak in phonon DOS) ABINIT LDA reproduces peak position, but absolute value at peak ~20% lower VASP GGA reproduces peak position and magnitude

Comparison of calculated and measured specific heat 9 Extension to Ag. Ga. S 2 and Ag. Ga. Te 2 lattice softening by Cu Ag replacement lattice softening by S Te replacement

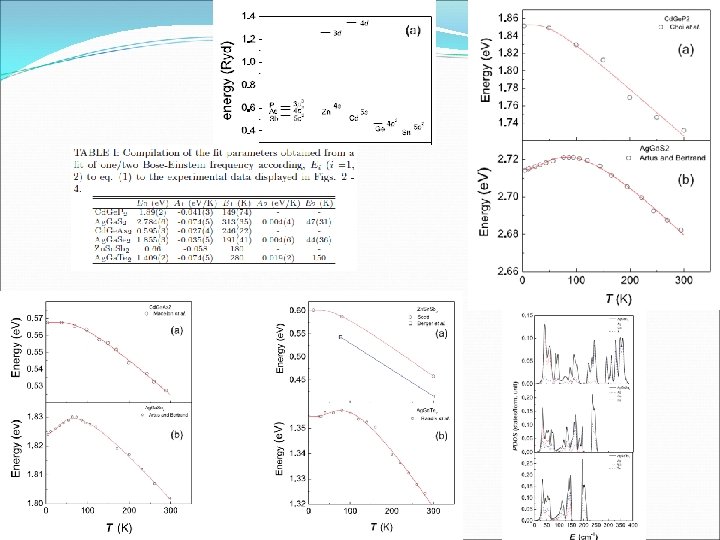
Even more properties? Inclusion of Temperature effects is computationally very expensive, e-ph interaction, … Many experimental results of T dependence of the gap in binary and ternary compounds. The degree of cation-anion hybridization on the electronic an vibrational properties, leads to anomalous dependence of the band gaps with temperature. The presence of d-electrons in upper VB lead to anomalies, like negative s-o splitting. For example in Cu or Ag chalcopyrite, the other constituents correspond to decrease the gap, but Cu or Ag tends to increase. The sum of boths effects generates a non monotonic dependence of gaps with T. It can be fitted using two Einstein oscillator according to: E 0 is the zero-point un-renormalized gap energy, A 1 is the contribution to the zero-point renormal. , n. B is the Bose-Eisntein function.

The admixture of p and d electrons in the valence bands produces anomalies e. g. in the temperature dependence of the energy gap: at low T the gap increases with T (up to~100 K) presumably because of the presence of delectrons. Above 100 K it decreases. Detailed theoretical explanation not yet available. Ag. Ga. S 2 Temperature dependence of the energy gap of Ag. Ga. S 2 with two-phonon fit. Cu. Ga. S 2 Temperature dependence of the energy gap of Cu. Ga. S 2 with two-phonon fit.

ELASTICITY Elasticity - deformation
")
σij = VOIGT’S NOTATION Cijkl εkl (only two index)


Some examples of elasticity under pressure


Mechanical stability criteria Pressure 0 GPa Born Criteria Pressure P ≠ 0 GPa Born Generalized Criteria
 (160 atoms")
YGa 5 O 12 garnet unit cell) (160 atoms

CONCLUSIONS: Ø Ab initio methods can provide interesting and useful information of the physics and chemistry of materials properties under high pressure, from small system to big systems. Phonons and elastic properties provide interesting info, dynamical and mechanical stability ØTemperature, S-O etc…T effects can be included. ØThese techniques can help to design and to understand problems in experimental interpretations. ØBut remember!!!!! WE USE APPROXIMATIONS

“An expert is a person who has made all the mistakes that can be made in a very narrow field”. Niels Bohr (1885 -1962)
")
Physics is to mathematics like sex is to masturbation. ” —Richard Feynman, (1918 -1988)

Thank you for your attention!
- Slides: 63