FMRP the protein product of the FMR 1

































- Slides: 37
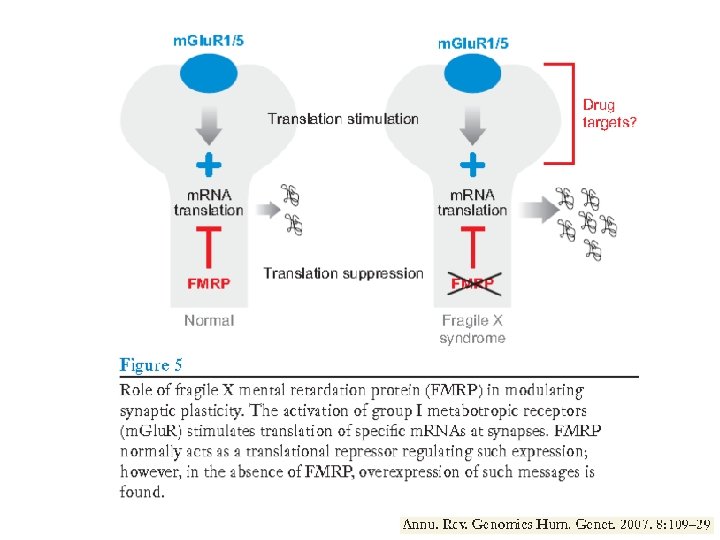
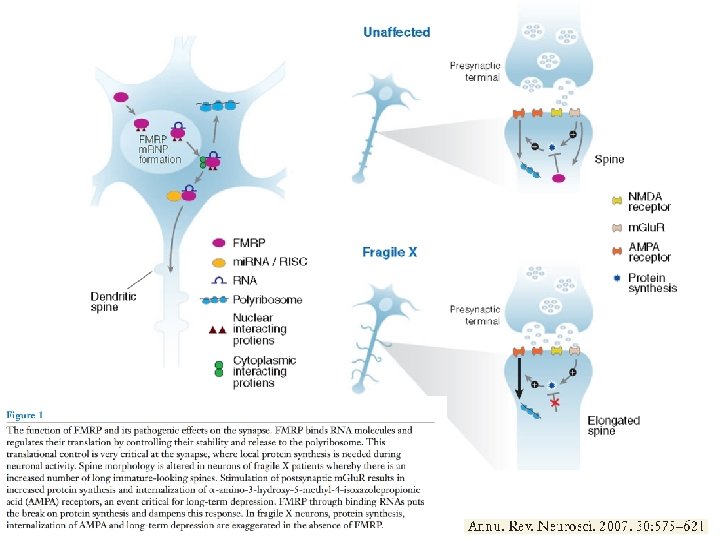
FMRP: the protein product of the FMR 1 gene • It is located largely in the cytoplasm but does make excursions to the nucleus. • It has 3 RNA binding domains; it associates with ribosomes and seems to be involved in translational regulation of a group of RNA targets. • The protein and its m. RNA localise in dendritic spines. • The protein binds to certain “target” RNA species and represses translation. • These are thought to be m. RNAs coding for proteins involved in neuronal development, synaptic transmission and cytoskeleton
i pazienti con X Fragile Ritardo mentale ritardo nel linguaggio comportamento autistico i bambini evitano lo sguardo, si infliggono autopunizioni alle mani fino a mordersi, ipersensibili agli stimoli ed iperattivi Postmortem examination of the brains of some fragile X patients has shown that the only consistent difference in their brain architecture abnormally developed dendritic spines of neurons, which are observed in different regions of the brain. When compared with the normal structures, those spines appear too long, thin and also slidely more numerous. Dendritic spines are the major sites of synaptic input one neuron gets from another and therefore it seems likely that this abnormality is the structural basis underlying mental impairment in fragile X patients. Abnormal dendritic spines have been also found in the FMR 1 KO mouse. patient unaffected
Malattie da triplette ripetute non codificanti
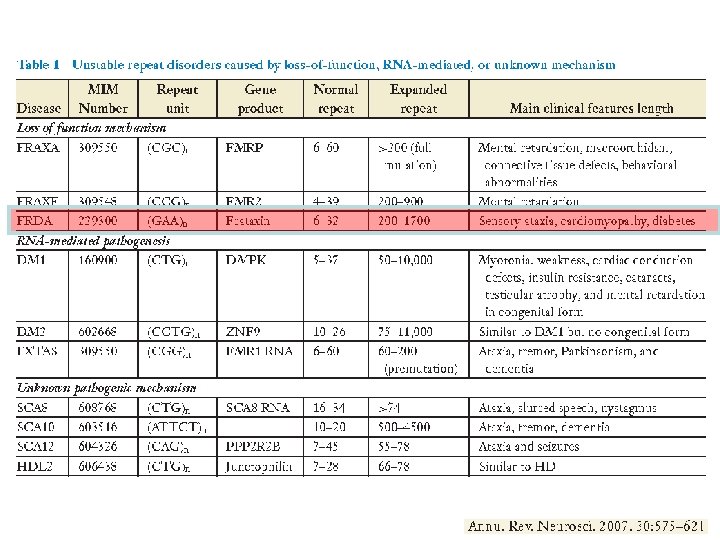
Friedreich’s Ataxia • The most common heritable form of ataxia associated with progressive gait and limb ataxia • Degeneration of large sensory neurons • Death is due to cardiac failure • Autosomal recessive • No anticipation • FRDA gene encoded frataxin • In FRDA disease the protein levels are severely reduced causing mitochondrial dysfunction
Friedreich’s Ataxia Fenotipo clinico La FA è una sindrome genetica trasmessa con modalità recessiva, caratterizzata da insorgenza precoce (prima della adolescenza) e decorso rapidamente progressivo. I primi sintomi generalmente appaiono nei bambini, ma l'esordio può essere variabile dall'infanzia alla età adulta. Perdita progressiva della coordinazione dei movimenti (sedia a rotelle nella III decade) Capacità della comunicazione compromessa dalla difficoltà ad articolare le parole (disartria) Diminuizione o assenza dei riflessi degli arti inferiori Perdita della sensibilità propriocettiva Perdita della vista in alcuni casi per atrofia dei nervi ottici e dell'udito Funzioni cognitive intatte
Friedreich’s Ataxia • The disease is caused by GAA repeat expansion • The repeat is located in intron 1, within an Alu sequences of the FRDA gene • Normal alleles 7 -34 repeats • Disease causing >100 repeats • GAG interruptions • Very low m. RNA levels
Friedreich’s Ataxia sindrome atassica + frequente in caucasici prevalenza circa 1: 50, 000 portatore 1: 120 gene FRDA proteina mitocondriale 210 aa espressa abbondantemente nel miocardio e midollo spinale e in tessuti ad elevato metabolismo come fegato reni e grasso bruno assente nella corteccia cerebrale enorme espansione del gene (I introne) inibisce la trascrizione interferendo con l'allungamento del trascritto
Friedreich’s Ataxia A differenza delle forme dominanti di atassia, la FA è dovuta a un deficit di energia imputabile al presunto ruolo della fratassina nella fosforilazione ossidativa mitocondriale e quindi oltre all'interessamento nervoso, si manifesta una cardiomiopatia ipertrofica che può portare nell'adulto a morte per insufficienza cardiaca o aritmia
Malattie da triplette ripetute non codificanti
Anticipazione nella distrofia miotonica grandmother has only late onset cataracts daughter has mild symptoms grandson has severe congenital myotonic dystrophy
DISTROFIA MIOTONICA Malattia di Steinert 1909 la più frequente distrofia muscolare autosomica incidenza 1: 8000 nati malattia multisistemica, interessando cuore, muscolatura liscia, SNC, occhio e ghiandole endocrine Esordio tardivo (> III decade) Quadro clinico vario tra grave e lieve
DISTROFIA MIOTONICA Fenotipo clinico malattia multisistemica con prevalente compromissione muscolare debolezza della contrazione muscolare e ritardo del rilassamento (miotonia) senso di rigidità muscolare Muscoli interessati: faccia e estremità
2 geni causali DMPK DM 1 19 q 13. 3. DMPK proteinchinasi 15 esoni DM 2 meno grave ZNF 9 tripletta I introne trascritta ma non tradotta fino a 5000 ripetizioni
Myotonic Dystrophy l Caused by CTG repeat in 3’ UTR of DMPK gene l There is a inverse correlation between CTG repeat size and age of onset l Anticipation occurs more dramatically in maternal transmissions l Late onset ¡ 50 - 100 repeats l Adult onset ¡ 100 – 1000 repeats l Congenital onset ¡ >1000 repeats l Tissue specificity probably caused by different rates of expansion l Caused by sequestering of proteins need for splicing of other genes by the expanded CUG tract in the m. RNA
Anticipation and Repeat Expansion in DM 84 late onset 112 205 160 adult onset congenital 2100 730 2100 Often resulting in congenital myotonic dystrophy in the later generations
Il caso della atrofia muscolare spino-bulbare e il recettore per gli androgeni
Gene AR: recettore degli androgeni AR: Xq 11 -q 12 Il recettore viene attivato dal DHT (diidrotestosterone) e stimola a sua volta la trascrizione di geni che favoriscono il differenziamento sessuale maschile All’interno di questo gene sono state rinvenute mutazioni di diversa natura: 1. Mutazioni con perdita di funzione: sindrome da insensibilità agli androgeni e sviluppo anatomico in senso femminile 2. Mutazioni con perdita parziale della funzione : varietà di stati sessuali ambigui
3. Espansione della tripletta CAG nell’esone 1 del gene : atrofia muscolare spinobulbare X-linked ad insorgenza tardiva. 4. Guadagno di funzione: la proteina è tossica per i neuroni: l’espansione modifica il dominio di attivazione della proteina 5. Normale variazione delle ripetizioni della tripletta CAG media la variazione della risposta agli androgeni: corte ripetizioni mediano una risposta forte e viceversa 6. ruolo nella suscettibilità alla calvizie maschile
Mutazioni dinamiche Espansioni imperfette di poli. A in sequenze codificanti
Altre malattie con “anticipazione” • bipolar affective disorder • schizophrenia • autism not associated with fragile X syndrome • spastic paraplegia • hereditary Parkinson’s disease
Anticipation by Shortening? l Anticipation also occurs in diseases caused by defects in telomere maintenance l Mutations in the genes encoding the RNA and the protein catalytic subunits of telomerase seen in conditions with anticipation l The telomeres undergo progressive shortening of the TTAGGG tract l After reaching a critical length this triggers apoptosis l Vulliamy T, (2004) Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nature Genetics 36 , 447 -449
Abstract Dyskeratosis congenita is an inherited syndrome characterised by mucocutaneous features, bone marrow failure, an increased risk of malig-nancy and other somatic abnormalities. There is a considerable range of clinical severity and in its occult form the disease may present as idiopathic aplastic anaemia. Genes responsible for X-linked, autosomal dominant and autosomal recessive forms of the disease have been identified and been found to encode products involved in telomere maintenance. Premature shortening of telomeres could account for the pathology, affecting the tissues that turn over most rapidly. However, the protein that is mutated in the X-linked disease, dyskerin, also plays a fundamental role in ribosome biogenesis, directing the pseudouridylation of ribosomal RNA using H/ACA small nucleolar RNAs as guides. Heterozygous mutations in the RNA component of telomerase (TERC) cause the autosomal dominant form of the disease through haploinsufficiency. Disease anticipation described in these families is associated with progressive telomere shortening through the generations. Heterozygous mutations in the reverse transcriptase component of telomerase (TERT) have a more variable role, often displaying incomplete penetrance and diverse clinical presentation. The autosomal recessive form of the disease is genetically heterogeneous, although one sub-type has been described in which NOP 10 is mutated. This small protein is also associated with the maturation of ribosomal RNA and the telomerase complex.
The DKC 1 gene provides instructions for making a protein called dyskerin. This protein is involved in maintaining structures called telomeres, which are found at the ends of chromosomes. Telomeres help protect chromosomes from abnormally sticking together or breaking down (degrading). In most cells, telomeres become progressively shorter as the cell divides. After a certain number of cell divisions, the telomeres become so short that they trigger the cell to stop dividing or to self-destruct (undergo apoptosis). Telomeres are maintained by two important protein complexes, telomerase and shelterin. Telomerase counteracts the shortening of telomeres by adding small repeated segments of DNA to the ends of chromosomes each time the cell divides. One component of telomerase, called h. TR, provides a template for creating the repeated sequence of DNA that telomerase adds to the ends of chromosomes. The dyskerin protein attaches (binds) to h. TR and helps stabilize the telomerase complex. In most types of cells, telomerase is either undetectable or active at very low levels. However, telomerase is highly active in cells that divide rapidly, such as cells that line the lungs and gastrointestinal tract, cells in bone marrow, and cells of the developing fetus. Telomerase allows these cells to divide many times without becoming damaged or undergoing apoptosis. Telomerase is also abnormally active in most cancer cells, which grow and divide without control or order. Dyskerin is also involved in the production of ribosomal RNA (r. RNA), a chemical cousin of DNA. Ribosomal RNA is required for assembling protein building blocks (amino acids) into functioning proteins.
v. Presymptomatic Testing: Testing for people who are genetically at risk for getting HD. v. Confirmatory Testing: Testing that determines whether people who are showing symptoms actually have HD. v. Prenatal Testing: Testing used to determine whether a fetus is at risk for HD.
v Usually includes sessions devoted to: genetic counseling, a neurological exam, a psychological interview, discussion of the results, and follow-up. v Neurological exam is meant to determine whether the patient has any symptoms, in which case they may choose to discontinue testing procedure. v Sessions are meant to ensure that the person about to undergo testing understands the implications of the knowledge of the results
Testing for HD • Presymptomatic testing available since 1980’s – Simple test since 1994 – ~16% of at-risk adults opt for testing • Geneticists do not recommend testing of at-risk children – Exceptions • Symptomatic children
v. It is usually strongly advised to bring a supportive friend to all testing sessions. v. It is not recommended to bring a sibling of someone else who is at risk for HD.
v. Accuracy of a positive or negative test result is almost 100% provided that another family member is known to have the gene for HD. v. Positive test results cannot predict when the symptoms will begin. v. Test results should always be confidential.
v. Personal relationships may change v. Reduction of uncertainty v. Preparation for the future v. Expenses v. Emotional trauma v. Discrimination