Fisiopatologa del Sistema Endocrino Fisiopatologa UNIBE Dr Jos

Fisiopatología del Sistema Endocrino Fisiopatología UNIBE Dr. José R. Fuchs C.

Alteraciones del Sistema Endocrino l l l DEFICIENCIA HORMONAL EXCESO DE HORMONAS RESISTENCIA DE ACCION A LAS HORMONAS l ESTADOS POR DEFICIENCIA HORMONAL: l Procesos destructivos l Infecciones ( Tuberculosis adrenal) l Infartos (Sheehan) l Inflamación ( DM secundaria a pancreatitis) l Tumores (células nulas de la pituitaria) l Autoinmune (tiroiditis de Hashimoto) l Hereditarias (enanismo pituitario) l EXCESO DE HORMONAS: l Sobreproducción de la glándula l Sobrestimulación de la glándula l Producida por tejido extraglandular l Iatrogénica l Destrucción de la glándula blanco

Alteraciones del Sistema Endocrino l RESISTENCIA HORMONAL l l PRODUCCION DE HORMONAS ANORMALES l l Generalmente de carácter hereditario l Deficiencia en receptores o post-recepción l Desarrollo de anticuerpos a las hormonas o sus receptores l Ausencia de células blanco Algunos tipos de DM Inmunoglobulinas que se fijan al receptor específico y lo estimulan Anticuerpos anti-receptores de insulina. DEFECTOS QUE AFECTAN SISTEMAS ENDOCRINOS MULTIPLES l l l Panhipopituitarismo Sindromes poliglandulares Sindromes de neoplasias endocrinas múltiples

HIPOTÁLAMO
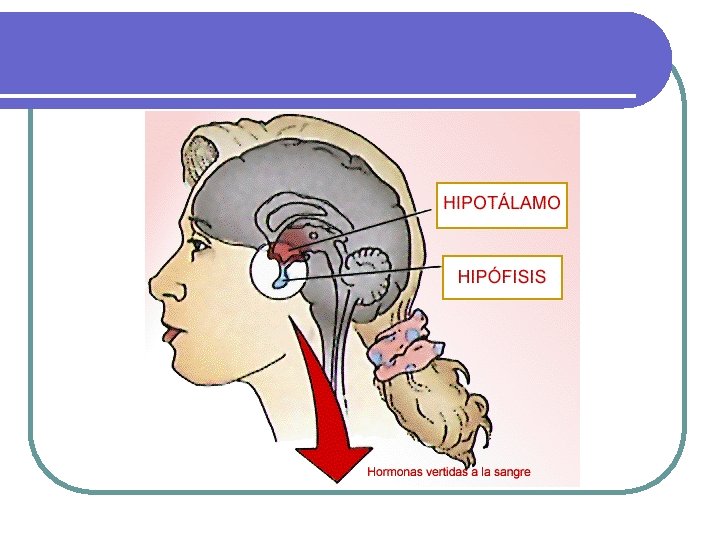
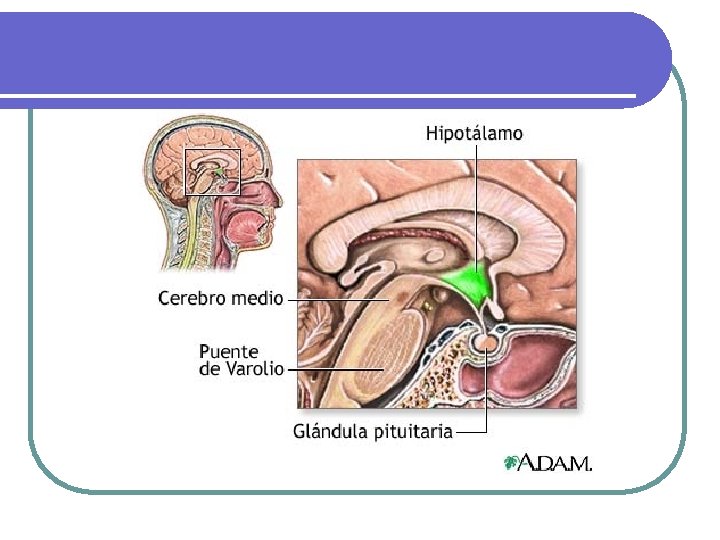

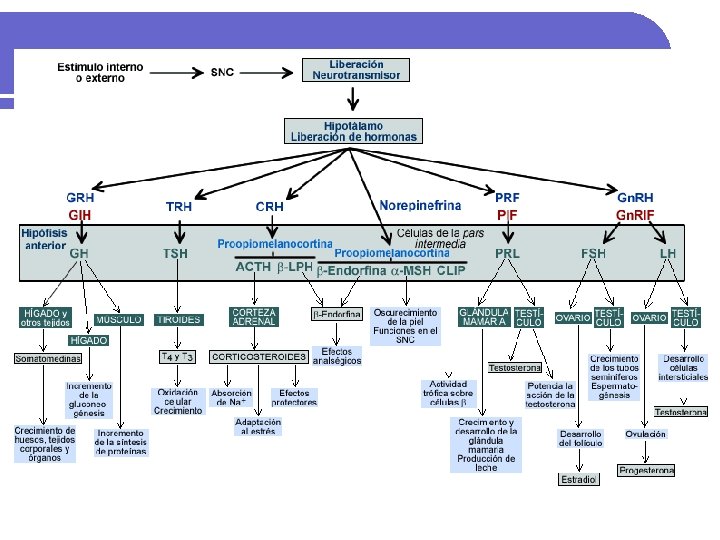
HIPOTÁLAMO l HIPOTALAMO l Peso: 4 g. Enfermedad ocurre sólo cuando el compromiso es bilateral. . l Funciones no endocrinas: l l l l Apetito y patrones alimentarios Regulación de la temperatura Ciclo sueño-vigilia Memoria y comportamiento Sed Función del sistema autonómico. Funciones endocrinas: l l Factores liberadores de hormonas estimulantes hipofisiarias: CRH; TRH; GHRH; Gn. RH. Dos sustancias inhibidoras: Somatostatina a la GH y la dopamina a la prolactina. Además de ocitocina y vasopresina u hormona antidiurética.

HIPOTÁLAMO l TUMORES. MANIFESTACIONES CLÍNICAS: l Tumores son de crecimiento lento y usualmente tienen que alcanzar gran tamaño para producir sintomatología: l l l l l Hidrocefalia por compromiso del tercer ventrículo Disfunción hipotalámica endocrina y no endocrina Hipopituitarismo parcial o total Demencia Trastornos en la ingesta de alimentos (obesidad, emación) Disfunción endocrina Los procesos agudos o de crecimiento rápido: Coma Alteraciones en la función autonómica

HIPOTÁLAMO l Lesiones en el hipotálamo anterior: Craneofaringioma Gliomas del nervio óptico Meningiomas Enf granulomatosas Germinomas Aneurismas de la carótida interna l Lesiones del hipotálamo posterior: Gliomas Hamartomas Ependimomas Germinomas Teratomas

Hipotálamo l l l l l CRANEOFARINGIOMAS 3 -5% de todas las neoplasias intracraneanas. Mayoría son supracelares, 15% intracelares. Frecuentemente quísticas y calcificadas. La mayoría se manifiestan en la infancia. 45% en > de 20 años y 20% en > de 40 años. CLINICA: Hipertensión endocraneana: cefalea, vómitos y papiledema. Trastornos visuales, hemianopsias. Baja estatura. En adultos el 80% tendrán trastornos visuales, cefalea, deterioro mental y cambios de conducta, hipogonadismo e hiperprolactinemia (50%). Menos frecuente: Diabetes insípida, obesidad, panhipopituitarismo.

HIPÓFISIS
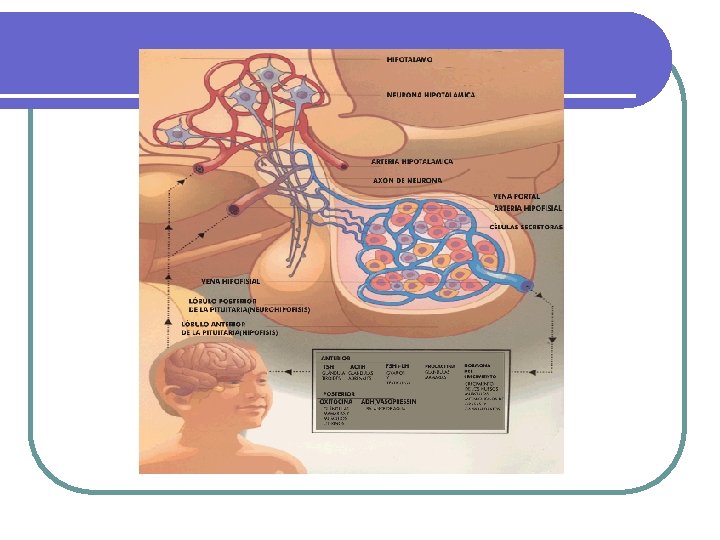
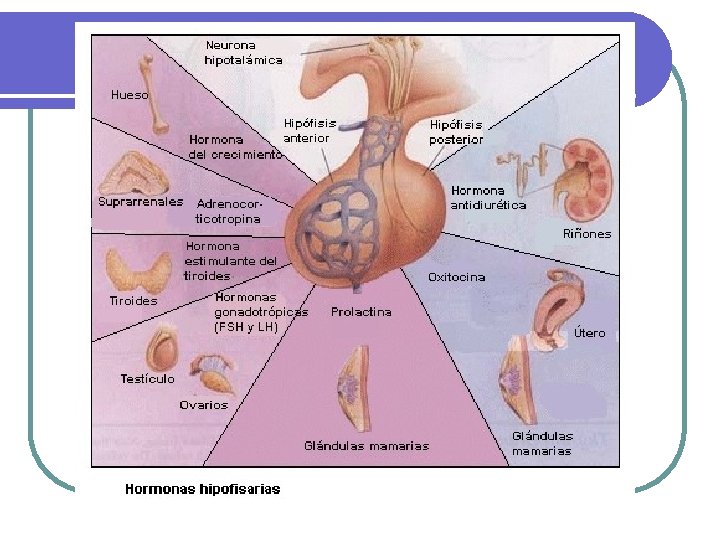

Hipófisis l TUMORES DE LA HIPOFISIS l 10 al 15% de todas los tumores intracraneanos l l Microadenomas < 1 cm diámetro. Macroadenomas > 1 cm. l Usualmente se diagnostican por: l Rx u otros estudios de imágenes de cráneo. l Defectos visuales, cefalea u otros defectos neurológicos, especialmente con macroadenomas l Datos de producción hormonal en exceso: hiperprolactinemia, cushing, acromegalia l Hipopituitarismo debido a destrucción de la región hipotálamo hipofisiaria. Apoplejía pituitaria. l

HIPÓFISIS l TIPOS DE TUMORES l l l l Porlactinoma: micro o macroadenoma. Secretores de HC: macroadenomas generalmente Enfermedad de Cushing: microadenomas productores de ACTH muy pequeños que a veces no se detectan por TAC O RM. Adenomas cromófobos, incidentalomas o de células nulas; no producen hormonas. (30 -40%) (los más frecuentes) MUY PERO MUY RAROS Secretores de (TSH). (EXCEPCIONALES) Secretores de (FSH y LH): hipogonadismo, problemas visuales y cefaleas. (MUY RAROS)

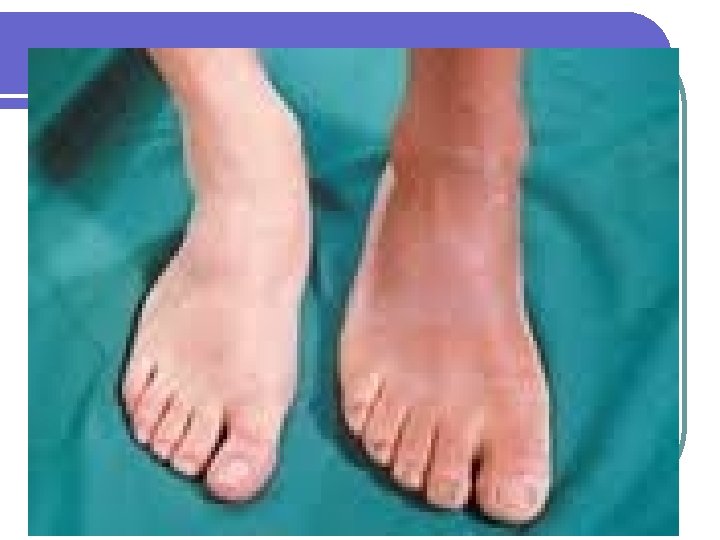
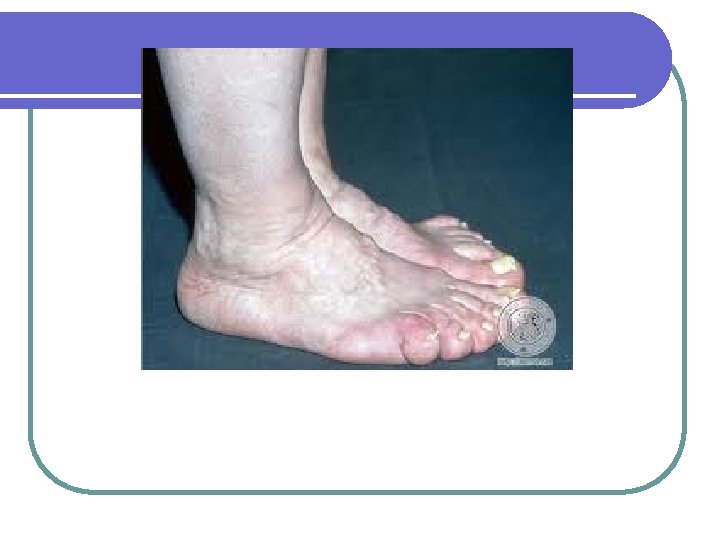
Adenomas hipofisiarios l Acromegalia: l l l l Exceso de hormona crecimiento Hipotálamo---GHRF est (+). Somatostatina inh (-) la producción de GH por la hipófisis : GH estimula la producción de somatomedinas (c) (ILGF) factor de crecimiento similar a la insulina, en el hígado Mayoría por macroadenomas hipofisiarios. 40 casos por millón. Raro por aumento de GHRF <1%. Producción ectópica de GH es rara: Carcinoma bronquial de células pequeñas Carcinoma medular de tiroides Carcinoides del intestino delgado

Crecimiento de las manos



CAUSAS DE HIPERPROLACTINEMIA l l l l ESTADOS FISIOLOGICOS: A Embarazo B Lactancia C Estrés E Estimulación del pezón F Ingesta de alimentos MEDICAMENTOS: A. Antagonistas de receptores dopaminérgicos l l l B Agentes que depletan dopamina l l Fenotiazinas Butirofenonas Tioxaminas Metoclopramida Sulpiride Metildopa Reserpina C Estrógenos D Opiáceos

Causas de hiperprolactinemia l ENFERMEDADES ASOCIADAS: l A Tumores pituirtarios: l l Prolactinomas Adenomas productores de GH y prolactina Adenomas secretores de ACTH y prolactina Adenomas cromófobos no funcionantes l B Enfermedad hipotalámica y del pedículo hipofisiario l l l Enfermedad granulomatosa (sarcoidosis) l Craniofaringiomas y otros tumores l Irradiación craneana l Sección del pedículo l Silla vacía l Anormalidades vasculares (aneurismas) l Hipofisitis linfocítica l Carcinoma metastásico C Hipotiroidismo primario D Insuficiencia renal crónica E Cirrosis F Trauma Tórax (cirugía, Herpes zoster) G Convulsiones l

Hipopituitarismo l CLASIFICACION DE HIPOPITUITARISMO: l l AISLADO: Deficiencia congénita de GNRH (hormona liberadora de gonadotrofinas). Deficiencia gonadal, ausencia de pubertad, bajos niveles plasmáticos de LH y FSH; se asocia a anosmia. Deficiencia aislada y adquirida de TSH: hipotiroidismo, pobre respuesta de TSH a TRH, se puede ver en la apoplejía pituitaria. Deficiencia aislada de ACTH, causando deficiencia selectiva de glucocorticoides, ausencia de hiperpigmentación, o contracción de volumen. El cortisol plasmático está bajo y no se eleva después de hipoglicemia inducida por insulina. Deficiencia aislada de hormona del crecimiento (GH). Estatura baja Todas estas deficiencias aisladas de las hormonas hipofisiarias son sumamente raras. l l

Hipopituitarismo l PANHIPOPITUITARISMO: l l Más común que las deficiencias aisladas. Destrucción de la pituitaria por un tumor, craniofaringioma o metastásico, tumores de la región hipotálamo-hipofisiaria. Síndrome de Sheehan o infarto pituitario post-parto: ausencia de lactación, bajos niveles de prolactina, ausencia de respuesta a TRH, insuficiencia gonadal, hipotiroidismo, insuficiencia suprarrenal y otras. Otras causas: enfermedades granulomatosas (ej. sacoidosis, tuberculosis, granulomatosis de Wegener), infecciones, radioterapia.

Fisiopatología de la glándula adrenal Fisiopatología UNIBE Dr. José R. Fuchs
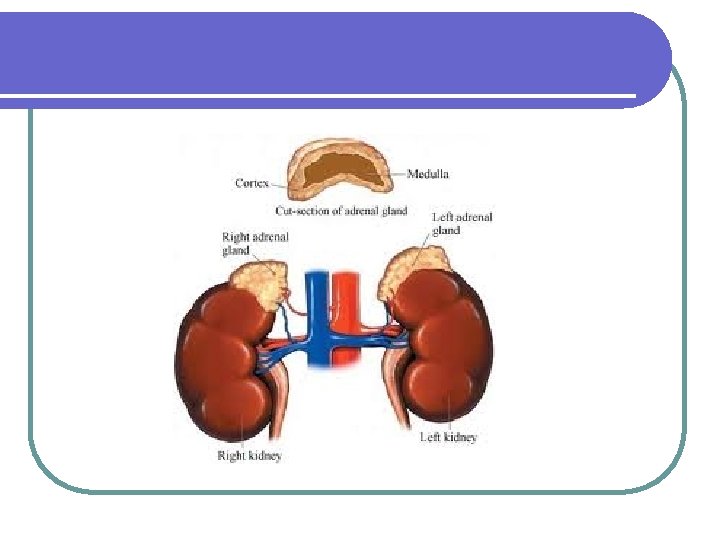
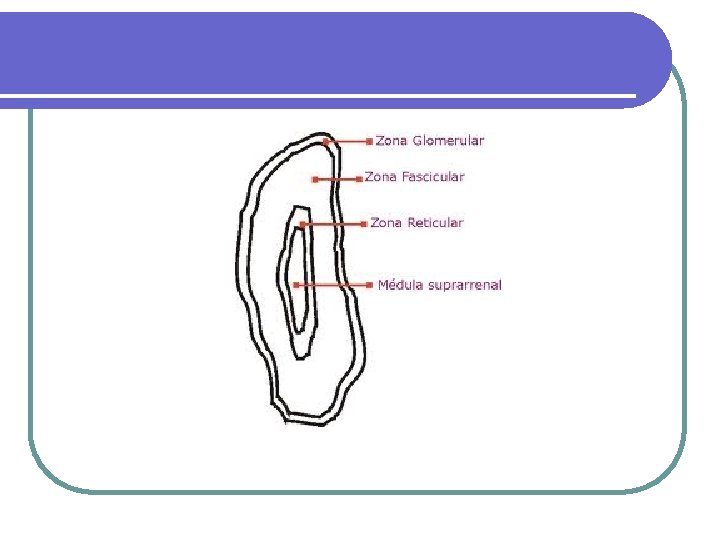
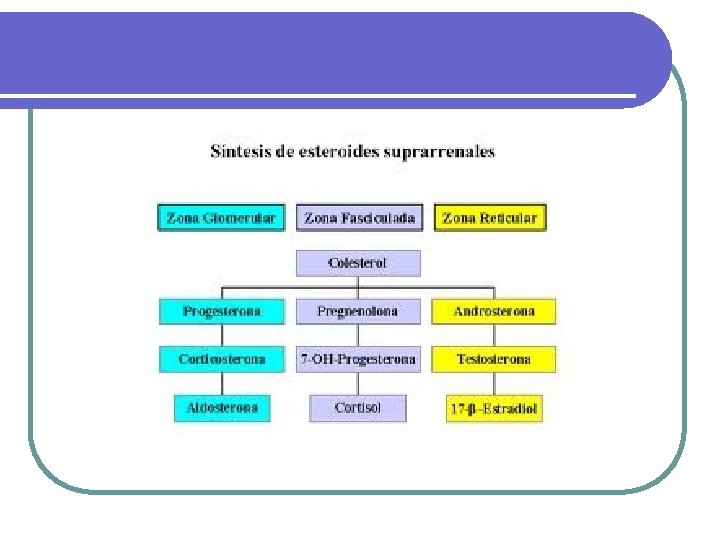

Síndrome de Cushing l La causa más común de Síndrome de Cushing es la iatrogénica debido a la administración de dosis terapéuticas de glucocorticoides. CAUSAS DE SINDROME DE CUSHING CAUSAS INCIDENCIA RELATIVA Dependientes de ACTH 75% Enfermedad de Cushing 60% Secreción ectópica de ACTH 15% Secreción ectópica de CRH Rara Independientes de ACTH Cáncer adrenal 25% Adenoma adrenal 10% Enfermedad adrenal micronodular Rara Ficticia o iatrogénica Muy común

Síndrome de Cushing TUMORES ASOCIADOS CON EL SINDROME DE ACTH ECTOPICO Carcinoma de células de avena 50% Tumores embrionarios 35% Carcinoma del timo Carcinoma islotes Carcinoma medular de la tiroides Carcinoide bronquial Feocromocitoma 5% Otros 10% Gonadas Carcinoma de la próstata y del cervix Tumores de origen desconocido

Enfermedad de Cushing l La enfermedad de Cushing (producción de ACTH por un microadenoma pituitario) es la causa más frecuente del Síndrome de Cushing. l Es 4 veces más frecuente en mujeres que en hombres y a menudo ocurre entre las edades de 20 a 40 años. l Usualmente es muy pequeño, sin embargo una TAC o una RM son capaces de detectarlos en un 50% de los casos.

Síndrome de Cushing Los tumores adrenales, causando un Síndrome de Cushing, se pueden presentar a cualquier edad, pero predominan en niños (particularmente carcinoma). l Los carcinomas son usualmente muy agresivos y grandes y pueden palparse en la mayoría de los casos; en contraste los adenomas son de crecimiento lento y de tamaño moderado (1, 5 a 6 cm de diámetro al diagnóstico), rara vez se asocian al MEN I. l La hiperplasia adrenal micronodular ocurre en niños y adultos jóvenes, la patogénesis bioquímica no se conoce con certeza, puede ocurrir esporádicamente como enfermedad familiar. l
: 1. Insuficiencia adrenal primaria a.")
INSUFICIENCIA ADRENAL CAUSAS DE INSUFICIENCIA SUPRARRENAL ETIOLOGÍA (OCURRENCIA %): 1. Insuficiencia adrenal primaria a. b. c. Autoinmune (70%) Tuberculosis (20%) Otras (10%) l SIDA Hemocromatosis l Infecciones por hongos Adrenoleucodistrofia l Hemorragia adrenal Neoplasia metastásica l Amiloidosis No respuesta congénita a la ACTH l Sarcoidosis l Hiperplasia adrenal congénita. 2. Insuficiencia adrenal secundaria a. b. c. Supresión adrenal por administración exógena de glucocorticoides o ACTH Secundario al tratamiento del Síndrome de Cushing Lesiones hipotalámicas o pituitarias.

INSUFICIENCIA ADRENAL l l l Las dos causas más comunes de insuficiencia adrenal son la adrenalitis autoinmune y la tuberculosis. En los países en vías de desarrollo, la TB puede ser más prevalente que la adrenalitis autoinmune. La Enfermedad de Addison autoinmune puede ser parte del Síndrome de deficiencia poliendocrina tipo I y II. La tipo I es una enfermedad de la niñez y se caracteriza por insuficiencia adrenal, hipoparatiroidismo, y candidiasis mucocutánea, también puede incluir hipogonadismo, anemia perniciosa, hepatitis crónica activa y alopecia. Tipo II (Síndrome de Schmidt) es una enfermedad de adultos jóvenes, se caracteriza por insuficiencia adrenal, enfermedad autoinmune de la tiroides, y DM tipo 1.
- Slides: 38