Facolt di Scienze Motorie Universit degli Studi di





 1.")
 1. 2. 3.")



 di membrana")
 Vantaggi ØSemplice ØEconomica ØBen accetta dal paziente ØPossibilità di intervenire")
 Svantaggi ØNon adatta per farmaci distrutti dall’acidità gastrica o dagli")

 nella circolazione sistemica ØEffetto rapido")



 Vantaggi ØAzione rapida: utile nelle situazioni")
 Svantaggi ØMaggior rischio di gravi effetti")
 Vantaggi ØAssorbimento rapido ØPossibilità di utilizzo nei pazienti incoscienti")
 Svantaggi ØProvoca talora dolore nella sede d’iniezione ØNon si")






 ØVagina")



 Ø Warfarin")





, quelli")

")





 quando un sito di")
 1. 4 milioni di polimorfismi a")







 Rilevanza clinica")

")



 e ipertermia maligna • • • Il gene")












 del rene varia con l’età.")





 DEI FARMACI Ø Rappresentano la quantità di farmaco attivo contenuta")
 DEI FARMACI Ø I livelli ematici di un farmaco dipendono")








 o gamma (g):")
: millesima parte del millilitro Ø Millilitro (ml): millesima")

- Slides: 98
Facoltà di Scienze Motorie Università degli Studi di Verona Corso di “Farmacologia” Lezioni n. 3 -4 (Farmacocinetica) Docenti: Guido Fumagalli e Roberto Leone
LA FARMACOLOGIA COMPRENDE: FARMACODINAMICA Studia i meccanismi d’azione dei farmaci e gli effetti biochimici e fisiologici degli stessi. FARMACOCINETICA Studia i movimenti del farmaco nell’organismo. FARMACOTERAPIA Studia l’impiego dei farmaci nella prevenzione e trattamento delle patologie. TOSSICOLOGIA Studia gli effetti nocivi dei farmaci e più in generale di qualsiasi sostanza chimica.
Fasi dell’azione farmacologica Somministrazione del farmaco • Disgregazione del composto • Soluzione dei principi attivi Farmaco disponibile per l’assorbimento • Assorbimento • Distribuzione • Escrezione Farmaco disponibile per l’azione I Fase farmaceutica Disponibilità farmaceutica II Fase farmacocinetica Disponibilità biologica Azione sui recettori III Fase farmacodinamica nei tessuti bersaglio Effetto
FARMACOCINETICA Studia i movimenti del farmaco nell’organismo. Le varie fasi della cinetica di un farmaco sono: Assorbimento Passaggio del farmaco dalla sede di applicazione al sangue attraverso le membrane biologiche Distribuzione del farmaco dal sangue ai diversi compartimenti dell’organismo Metabolismo o Biotrasformazione Modificazioni chimiche il farmaco subisce nell’organismo, principalmente ad opera del fegato Eliminazione del farmaco dall’organismo, prevalentemente avviene ad opera del rene
ASSORBIMENTO L’entità e la velocità di assorbimento di un farmaco dipendono essenzialmente dalla: 1. 2. 3. 4. Via di somministrazione Forma farmaceutica Liposolubilità del farmaco Per la via orale dal p. H dell’ambiente e dalla costante di dissociazione del farmaco (p. Ka)
Principali vie di somministrazione dei farmaci Enterali (in cui si utilizza l’apparato gastroenterico) 1. Orale 2. Sublinguale 3. Rettale
Principali vie di somministrazione dei farmaci Parenterali (diverse rispetto all’apparato gastroenterico) 1. 2. 3. 4. 5. 6. 7. Endovenosa Intramuscolare Sottocutanea Intradermica Inalatoria Intratecale Intrarteriosa
Principali vie di somministrazione dei farmaci Topiche 1. 2. Percutanea Transmucosa
Ordine decrescente delle principali vie di somministrazione in relazione alla velocità ed entità dell’ASSORBIMENTO 1. Endovenosa (non c’è la fase di assorbimento) 2. Inalatoria 3. Sublinguale 4. Sottocutanea 5. Intramuscolare 6. Intradermica 7. Rettale 8. Orale
Schema della morfologia strutturale delle membrane cellulari Proteine Canali Code molecolari lipofile Teste molecolari idrofile
Assorbimento attraverso carrier (trasportatore) di membrana
Via Orale (per os) Vantaggi ØSemplice ØEconomica ØBen accetta dal paziente ØPossibilità di intervenire in caso di errore ØUtile nelle terapie protratte
Via Orale (per os) Svantaggi ØNon adatta per farmaci distrutti dall’acidità gastrica o dagli enzimi digestivi ØPossibile interazione con il cibo ØAssorbimento variabile sia come entità che velocità ØEffetto del primo passaggio epatico ØInadatta per soggetti non cooperanti (neonati, pazienti incoscienti, ecc. ) o che vomitano ØNon indicata nei casi in cui si vuole un effetto immediato
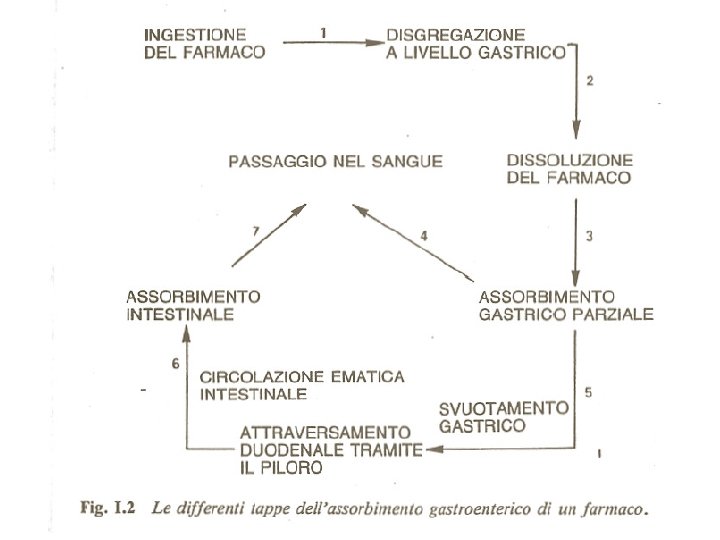
Schema della circolazione sanguigna dell’app. gastroenterico
Via Sublinguale Vantaggi ØPassaggio diretto (evitando il filtro epatico) nella circolazione sistemica ØEffetto rapido ØUtile per l’autosomministrazione al bisogno ØPossibilità di interrompere l’effetto sputando la compressa
Via Sublinguale Svantaggi ØIrritazione della mucosa ØInadatta per uso regolare e frequente ØNecessità di evitare la deglutizione
Via Rettale Vantaggi ØUtile nel caso di vomito, paziente incosciente, bambini piccoli ØPer somministrazione di farmaci irritanti per lo stomaco ØPer azione locale (es. purganti irritativi) ØSi evita in parte il filtro epatico
Via Rettale Svantaggi ØMateriale fecale può interferire con l’assorbimento ØPossibile irritazione locale ØNon particolarmente gradita dai pazienti
Via Endovenosa (e. v. o i. v. ) Vantaggi ØAzione rapida: utile nelle situazioni di emergenza e per ottenere un’elevata concentrazione di farmaco nel sangue ØPrecisione nel dosaggio ØPossibilità di somministrare volumi notevoli ØPossibilità di somministrare sostanze irritanti ØUtilizzabile nei pazienti incoscienti
Via Endovenosa (e. v. o i. v. ) Svantaggi ØMaggior rischio di gravi effetti avversi (flebiti, embolie, shock anafilattico, infezioni) ØNel caso di errore (dosaggio o forma farmaceutica non idonea) non è possibile evitare gli effetti avversi provocati ØNon particolarmente gradita dai pazienti
Via Intramuscolare (i. m. ) Vantaggi ØAssorbimento rapido ØPossibilità di utilizzo nei pazienti incoscienti ØPiù sicura rispetto alla via e. v. ØAdatta anche per preparati deposito (ritardo)
Via Intramuscolare (i. m. ) Svantaggi ØProvoca talora dolore nella sede d’iniezione ØNon si possono somministrare farmaci necrotizzanti
Via Inalatoria Vantaggi ØAssorbimento estremamente rapido ØSi evita il filtro epatico ØPossibilità di autosomministrazione ØUtile anche per azione locale ØAdatta per somministrazione di gas (anestetici)
Via Inalatoria Svantaggi ØMinor controllo del dosaggio ØNecessità di apparecchiature particolare ØPossibilità di irritazione locale
Via Sottocutanea Vantaggi ØAzione rapida ØAdatta per preparati deposito ØPossibilità di autosomministrazione
Via Sottocutanea Svantaggi ØNon adatta per sostanze irritanti ØAssorbimento scarso in pazienti con insufficienza circolatoria periferica ØIniezioni ripetute possono provocare lipoatrofia conseguente scarso assorbimento
Via Percutanea Ø Utilizzata generalmente per un’azione locale a livello della cute stessa o del derma, talvolta si utilizza anche per ottenere un effetto sistemico (ad esempio con i cerotti transdermici). Per avere o aumentare questo effetto è possibile fare delle manipolazioni: l’occlusione prolungata della superficie cutanea con plastica risulta più efficace dell’operazione di stripping con nastro adesivo (cioè l’eliminazione dello strato corneo), e la combinazione dei due metodi ha un effetto sinergico. Ciò permette di raggiungere concentrazioni di farmaco superiori a quelle misurabili dopo somministrazione orale. Bisogna considerare che la diffusione in profondità avviene a forma di cono con la base verso l’alto e perciò per avere una esteso interessamento a livello di muscolatura sottocutanea è necessario applicare sulla epidermide il farmaco in maniera estesa. L’assorbimento cutaneo di farmaci varia a seconda della zona corporea: è massima per cuoio capelluto, fronte, mandibola, ascella, scroto è minima per palmo, caviglia, arco plantare.
Via Percutanea Ø Se la cute è lesionata possono essere assorbiti farmaci altrimenti non assorbibili o si può aumentare l’assorbimento. Ø Forma particolare di somministrazione per via cutanea è la ionoforesi: cioè l’impiego di corrente elettrica continua che favorisce l’assorbimento del farmaco.
Altre comuni sedi di somministrazione di farmaci per azione locale ØNaso (gocce, spray) ØVagina (ovuli, candelette, irrigazioni, creme) ØOrecchio (gocce) ØOcchio (colliri, pomate, bagni oculari)
FATTORI CHE POSSONO MODIFICARE L’ASSORBIMENTO DEI FARMACI Via Orale ØInterferenza con il cibo (vedi diapo successive) ØDiarrea (aumentata peristalsi intestinale) ØVomito ØInterazione tra farmaci ØCondizioni di malassorbimento (anziani) ØResezioni gastriche o intestinali ØStenosi pilorica
Farmaci e cibo Ø In generale la somministrazione di un farmaco per os lontana dai pasti comporta un assorbimento più rapido e completo. Ø La somministrazione in vicinanza dei pasti può limitare i fenomeni irritativi alle mucose. Ø Alcuni farmaci possono interagire con determinati alimenti, ad esempio le tetracicline (antibatterici) si legano al calcio contenuto nel latte (o formaggi) e questo impedisce il loro assorbimento. Ø Per somministrazione prima dei pasti si intende: da 30 a 0 minuti prima del pasto Ø Per somministrazione dopo i pasti si intende: entro 30 minuti dopo il pasto Ø Per somministrazione lontano dai pasti si intende: 3 -4 ore prima o dopo il pasto
============================== Effetto della contemporanea assunzione di cibo sull'assorbimento di alcuni farmaci somministrati per via orale ============================== Assorbimento ridotto Assorbimento aumentato ----------------------------------------------------Ampicillina Griseofulviana Amoxicillina Carbamazepina Rifampicina Propranololo Aspirina Metoprololo Isoniazide Spironolattone Levodopa Idralazina ==============================
Esempi di interazioni con alimenti Ø Tetracicline + alimenti (es. latte, formaggi) Ø Warfarin + succo di mirtillo Ø Diversi farmaci + succo di pompelmo (vedi oltre) Ø Farmaci che agiscono sul SNC + alcool
FATTORI CHE POSSONO MODIFICARE L’ASSORBIMENTO DEI FARMACI Via Parenterale ØEdemi e ascessi (via s. c. ) ØInsufficienza circolatoria periferica (i. m. , s. c. ) ØShock e fuoriuscita del farmaco dalla vena (e. v. ) ØInterazione tra farmaci (vasocostrittori, vasodilatatori)
DISTRIBUZIONE DEI FARMACI NELL’ORGANISMO Il processo di distribuzione di un farmaco dal sangue ai diversi compartimenti dell’organismo è influenzato da diversi fattori: Ø Caratteristiche chimico-fisiche del farmaco (in particolare la sua liposolubilità) Ø Vascolarizzazione degli organi (un farmaco raggiunge più velocemente gli organi maggiormente perfusi dal sangue, quali cuore, encefalo, fegato e rene, ricevono il farmaco) Ø Percentuale di farmaco legato alle proteine plasmatiche Ø Presenza di particolari strutture anatomico/funzionali (barriera placentare, barriera emato-encefalica)
Siti di deposito cellulare Ø I farmaci possono legarsi anche con costituenti cellulari tissutali quali proteine, fosfolipidi, nucleoproteine. Ø Si possono così avere dei siti di deposito a livello di alcuni tessuti nei cui confronti un farmaco ha un particolare TROPISMO Ø Esempi di tropismo: §Tetracicline (antibatterici) verso il tessuto osseo §Tiopentale (anestetico) verso il tessuto adiposo §Clorochina (antimalarico) verso il fegato §Amiodarone (antiaritmico) verso la tiroide
METABOLISMO O BIOTRASFORMAZIONE DEI FARMACI Ø Con il termine metabolismo si intendono le modificazioni chimiche un farmaco subisce nell’organismo. Ø Sede principale dei processi metabolici è il FEGATO per l’azione degli enzimi microsomiali delle cellule epatiche. Ø Altre sedi di metabolizzazione di minore importanza sono il rene, il polmone, l’intestino (anche per azione della flora batterica).
METABOLISMO O BIOTRASFORMAZIONE DEI FARMACI Ø Le reazioni chimiche con le quali si attua il metabolismo dei farmaci sono: OSSIDAZIONE RIDUZIONE IDROLISI FASE I (Citocromo P 450) CONIUGAZIONE FASE II Ø Pazienti con patologie epatiche possono avere dei problemi di metabolizzazione di farmaci.
Fase I Gli enzimi della fase I predominano nel reticolo endoplasmatico liscio (microsomi), quelli della fase II nel citosol
METABOLISMO O BIOTRASFORMAZIONE DEI FARMACI Ø Le caratteristiche lipofile, che promuovono il passaggio dei farmaci attraverso le membrane biologiche e il conseguente accesso ai siti d’azione, ostacolano la loro eliminazione dall’organismo. Ø La biotrasformazione dei farmaci ha un’importanza fondamentale per la cessazione della loro attività biologica e per l’eliminazione dall’organismo. Ø Generalmente le reazioni di biotrasformazione danno origine a composti più polari, metaboliti inattivi che vengono più facilmente escreti dall’organismo.
LE QUATTRO POSSIBILITÀ DI BIOTRASFORMAZIONE DEI FARMACI Farmaco attivo Metabolita inattivo (caso più frequente) Farmaco inattivo (profarmaco) Metabolita attivo Farmaco attivo Metabolita tossico
ALCUNI ESEMPI DI PROFARMACI Ø LEVODOPA Utilizzata nel morbo di Parkinson è il precursore inerte della dopamina. La conversione metabolica (decarbossilazione) avviene nel SNC, principalmente entro i terminali presinaptici dei neuroni dopaminergici nello striato. Nella pratica clinica la levodopa viene somministrata assieme alla carbidopa o alla benserazide, inibitori periferici della decarbossilasi, per impedire che venga inattivata prima di raggiungere il SNC. Ø CODEINA Analgesico oppiaceo (contenuto nell’oppio) che esplica la sua azione antidolorifica dopo trasformazione nell’organismo in morfina. Ø ENALAPRIL-QUINAPRIL-FOSINOPRIL-RAMIPRIL ACE-inibitori che diventano attivi quando convertiti, dalle esterasi epatiche, rispettivamente a enalaprilato, quinaprilato, fosinoprilato, ramiprilato.
SISTEMA CITOCROMO P 450 MONOOSSIGENASI Ø E’ costituito da proteine di membrana, contenenti un gruppo eme, localizzate nel reticolo endoplasmatico liscio, prevalentemente a livello epatico. Producono una caratteristica banda di assorbimento spettrofotometrico a 450 n. M. Ø La famiglia del gene P 450 (CYP) si è differenziata (in miliardi di anni) garantendo il metabolismo di un numero sempre crescente di composti chimici ambientali, tossine alimentari, farmaci. Ø La superfamiglia di enzimi che ne è derivata catalizza una varietà enorme di reazioni (ossidazione, riduzione) nei confronti di diversi substrati, differenti dal punto di vista chimico. Ø A seconda della somiglianza nella catena di aminoacidi gli isoenzimi sono raggruppati in famiglie e subfamiglie. Attualmente nell’uomo conosciamo 18 famiglie di CYP 450, con 42 sottofamiglie e 57 geni codificanti.
Nomenclatura dei citocromi P 450, esempio: CYP 2 D 6 Ø CYP = citocromo P 450 Ø 2 = famiglia Ø D = sub-famiglia Ø 6 = specifico isoenzima (specifico gene) La nomenclatura è basata sui geni e non ha implicazioni funzionali
Percentuale dei farmaci metabolizzati dagli enzimi appartenenti alla classe del citocromo P 450 CYP 3 A 4 (36%, secondo alcune stime potrebbero superare il 50%) CYP 2 D 6 (19%) CYP 2 C 19 CYP 2 C 9 CYP 1 A 2 CYP 2 E 1 CYP 2 B 6 CYP 2 A 6
PRINCIPALI FATTORI RESPONSABILI DELLA VARIABILITÀ NEL METABOLISMO DEI FARMACI Ø Polimorfismi genetici (variazioni a livello dei geni presenti in >1% popolazione) Ø Stati fisiologici (età, sesso) Ø Stati patologici Ø Induzione o inibizione da farmaci concomitanti o fattori ambientali
Polimorfismi Genetici SNPs Si parla di Single Nucleotide Polymorphism (SNPs) quando un sito di un gene presenta diversi nucleotidi (e la proteina diversi aminoacidi) in individui della stessa specie. Aplotipo Set di SNPs correlati tra loro che vengono ereditati assieme
Basi molecolari della variabilità umana (Sachinanandan, Nature 2001) 1. 4 milioni di polimorfismi a singolo nucleotide n 60. 000 in zone codificanti n
Polimorfismi Genetici Farmacocinetica Assorbimento Farmacodinamica Recettori Distribuzione Metabolismo Escrezione Canali ionici Enzimi Sistema immunitario TRENDS in Pharmacological Sciences, 2001, 22 : 298 -305
Effetto dei polimorfismi: -Farmacocinetica Metabolismo -Farmacodinamica Efficacia/Tossicità Evans et al. , NEJM 2003; 348: 538 -549
Esempi di polimorfismi enzimatici non legati al citocromo P 450 Deficit della pseudocolinesterasi o pseudocolinesterai atipiche § Apnea da succinilcolina Deficit della Glucosio-6 -Fosfato Deidrogenasi (G 6 PD) § Anemia emolitica da antimalarici o da altri farmaci ossidanti Carenza di metaemoglobina reduttasi § Metaemoglobinemia da clorochina Carenza di glutatione § Epatotossicità da paracetamolo Acetil-tranferasi (acetilatori lenti) § Neuropatie da isoniazide
Esempio di polimorfismo a livello di citocromo P 450 Gasche Y et al. , NEJM, 2004, 351: 282731 Case report: Perdita di coscienza (score 6 nella scala del coma di Glasgow) - CYP 3 A 4: responsabile dell’ 80 % del metabolismo della codeina (inibito da voriconazolo e claritromicina) - CYP 2 D 6: ultrarapid metabolism responsabile del 10% del metabolismo di codeina -Insufficienza renale acuta per accumulo glucuronidi 75% della codeina totale veniva trasformato in morfina e rispettivi metaboliti rispetto al 10% dell’individuo normale
ATP GLICOPROTEINA-P n n n Pompa di efflusso multifarmaci energia-dipendente Fosfoglicoproteina di surperfice Trovata nell’intestino, rene, fegato, cervello, testicoli, placenta, surrenali Ambudkar SV et al. , Ann Rev Pharmacol Toxicol 1999; 39: 361 -98
• riduce ASSORBIMENTO GI • riduce DISTRIBUZIONE • aumenta ELIMINAZIONE Intestino GLICOPROTEINA-P e FARMACOCINETICA Spazio intravascolare Spazio Interstiziale
Influenza della Variabilità genetica della p-Gp sulla farmacocinetica Evans et al. , NEJM 2003; 348: 53849
Polimorfismi genetici di alcuni recettori e loro rilevanza clinica Recettore (difetto genetico) Rilevanza clinica Androgeni Risposta alla terapia antiandrogenica Recettore delle sulfuniluree (SUR) Iperinsulinismo Infantile, diabete (1) β 2 -Adrenocettori (mutazioni missense) Alterata risposta ad antiasmatici; mortalità nell’insufficienza cardiaca Canali del potassio (mutazioni missense) Sindrome del QT lungo (LQTS) Canali del sodio (mutazioni missense) Sindrome del QT lungo (LQTS) Modified from Weber, Mut Res 2001; 479: 1
Variabilità genetica e risposta agli agonisti β 2 Evans et al. , NEJM 2003; 348: 538 -
Sindrome del QT Lungo (LQTS)
Canali e repolarizzazione K+ K+ Na+
Un rallentamento eccessivo della fase 3 può portare all’insorgenza di un potenziale d’azione precoce. Ciò determina un’aumento del rischio di Torsioni di Punta (Td. P) che possono interrompersi o dar luogo a fibrillazione ventricolare con l’aumento del rischio rispettivamente di sincope o morte improvvisa Habriel H et al. , Swiss Med Wkly 2004, 134; 685 -694
IPERTERMIA MALIGNA • • Raro disturbo del muscolo scheletrico, potenzialmente fatale, che può essere scatenato dall’uso di anestetici volatili e miorilassanti depolarizzanti (~ sindrome maligna da neurolettici) Caratterizzata clinicamente da febbre, contratture muscolari, tachicardia, aumento della CO 2 espiratoria, lattacidosi e ipertermia Il gene ryr 1 sul cromosoma 19 q 13. 1 (locus MSH-1) è associato a più del 50% di tutte le forme familiari di ipertermia maligna (Mc. Carthy et al. , 2000) La mutazione R 614 C della proteina codificata da ryr 1 è responsabile dell’ipertermia maligna umana (Gillard et al. , 1991)
Recettore alla rianodina (Ry. R 1) e ipertermia maligna • • • Il gene ryr 1 codifica per un canale al Ca 2+ a larga conduttanza (Ry. R 1) che media il rilascio di Ca 2+ dal reticolo sarcoplasmatico Le mutazioni di Ry. R 1 determinano un’aumentata sensibilità delle fibre muscolari scheletriche agli stimoli che attivano il rilascio di Ca 2+ attraverso tale recettore Tale fenomeno determinerebbe un abnorme aumento del Ca 2+ citosolico e un’iperattivazione del metabolismo muscolare aerobico e anaerobico
INTERAZIONI TRA FARMACI Ø Le interazioni tra farmaci possono verificarsi a diversi livelli influenzando la farmacocinetica o la farmacodinamica dei farmaci stessi. Le interazioni conosciute sono moltissime tuttavia quelle di rilevanza clinica maggiore (da ricordare) sono relativamente poche. Ø Le interazioni più frequenti sono quelle a livello del metabolismo dei farmaci, dovute a meccanismi di induzione o inibizione enzimatica Ø Alcune volte le interazioni tra farmaci possono essere sfruttate per avere un maggiore effetto terapeutico o per contrastare fenomeni di intossicazione. Nella maggioranza dei casi, tuttavia, le interazioni sono alla base della comparsa di reazioni avverse. Ø I pazienti vanno educati a non aggiungere farmaci (ad esempio per autoprescrizione) alla terapia prescritta dal medico, in modo da evitare interazioni tra farmaci.
Interazioni tra farmaci Ø Sommazione Farmaco A Farmaco B Meccanismo A Meccanismo B Effetto finale = Effetto A + Effetto B Ø Addizione Farmaco A Farmaco B Meccanismo comune Effetto finale = Effetto (A+B)
Interazioni tra farmaci Potenziamento Farmaco A • Assorbimento Sinergismo Farmaco A • Eliminazione • Spiazzamento dall’accettore Farmaco B • Inibizione enzimatica Recettore Effetto finale > effetto A Farmaco B Risposta terapeutica Effetto finale > effetto A + effetto B
Interazioni tra farmaci Ø Antagonismo fisiologico Farmaco A Farmaco B Recettore A Recettore B Effetti opposti a quelli di A Effetto finale < effetto A Ø Ø Antagonismo recettoriale Farmaco A (agonista) Farmaco B (antagonista) Recettore Effetto finale < effetto A Antagonismo Chimico = farmaco A neutralizza chimicamente farmaco B*
Interazioni tra farmaci Ø Degradazione Ø Neutralizzazione Farmaco A Assorbimento Ø Eliminazione Ø Induzione enzimatica Ø Farmaco A + Farmaco B Inattivazione Chimicofisica Farmaco B Recettore Effetto finale < effetto A Effetto finale assente
INIBIZIONE ENZIMATICA Ø I farmaci che sono substrato dello stesso enzima possono inibire reciprocamente il loro metabolismo, ma spesso non ad un livello clinicamente rilevante. Ø Il meccanismo più comune di inibizione enzimatica è il legame competitivo a un citocromo P 450 (CYP), tuttavia alcuni farmaci inibiscono l’attività enzimatica senza essere substrato dell’enzima. Ø La potenza dell’inibizione può essere più importante del suo meccanismo. Il ketoconazolo e l’itraconazolo, ad esempio, possono inibire quasi completamente il CYP 3 A 4 anche a concentrazioni molto basse. Anche l’eritromicina è un potente inibitore del CYP 3 A 4, ma per un motivo differente; si lega con legame covalente all’enzima e lo inattiva in modo che l’effetto persista anche dopo che il farmaco è stato eliminato. Ø L’inibizione del metabolismo epatico inizia non appena nel fegato vi siano concentrazioni sufficienti dell’inibitore (in genere dopo poche ore dall’assunzione). L’effetto dell’inibizione sul metabolismo di un altro farmaco perciò è usualmente massimo nelle prime 24 ore. Ø Tuttavia , nonostante che l’inibizione insorga rapidamente, la comparsa dell’effetto clinico conseguente (generalmente una reazione tossica) può essere più ritardata. Ø L’inibizione enzimatica generalmente termina più rapidamente rispetto all’induzione enzimatica.
INDUZIONE ENZIMATICA Ø Un aumento dell’attività degli enzimi metabolizzanti che determina una riduzione dei livelli serici di un dato farmaco, è generalmente dovuta alla stimolazione della sintesi dell’enzima (da parte degli induttori enzimatici). Ø Gli induttori enzimatici stimolano il metabolismo di altri farmaci in maniera graduale. Sebbene l’effetto dell’induzione può essere individuato anche entro i primi due giorni di terapia, generalmente occorre una settimana prima che l’effetto massimo compaia. Ø Il tempo di comparsa del fenomeno dell’induzione dipende comunque anche dall’emività del farmaco inducente. Ad esempio la rifampicina, che ha una emivita relativamente breve, induce gli enzimi più rapidamente del fenobarbitale (induttore con emivita più lunga). Al contrario l’effetto dell’induzione si protrarrà più a lungo se determinata da un induttore con emivita più lunga.
INIBITORI E INDUTTORI DEL METABOLISMO DI FARMACI • INIBITORI • Amiodarone • Antimicotici imidazolici • Alcuni farmaci anti-HIV • INDUTTORI • Cimetidina • Carbamazepina • Macrolidi (NO azitr. ) • Fenobarbital • Isoniazide • Fenitoina • Fluorochinoloni • Rifampicina/rifabutina • Alcuni SSRI • Erba di San Giovanni (iperico) • Inibitori pompa proton. • Chinidina • Succo di pompelmo
Succo di pompelmo Il succo di pompelmo, ma non quello d'arancia dolce, aumenta la biodisponibilità di diversi farmaci, in particolare dei Ca-antagonisti. Nel caso della felodipina, che normalmente ha una biodisponibilità del 15% dopo metabolismo di primo passaggio, il succo di pompelmo produce concentrazioni di farmaco circa 3 volte più elevate della norma. Le conseguenze nei pazienti ipertesi borderline sono un'aumentata riduzione della pressione arteriosa ed un incremento della frequenza cardiaca. Le reazioni avverse correlate alla vasodilatazione (es. cefalea) sono di conseguenza più frequenti. Il succo di pompelmo inibisce selettivamente, nel tratto GI, il CYP 3 A 4. L'interazione tra felodipina e succo di pompelmo chiarisce due importanti concetti: l'importanza dell'intestino come sede di farmacometabolismo e che l'interazione dipende dalla via di somministrazione del farmaco. (Il succo di pompelmo non interagisce con farmaci somministrati per via endovenosa).
Succo di pompelmo • La durata dell'inibizione intestinale del CYP 3 A 4 dura fino a 24 ore dopo l'assunzione del succo. Così anche se si ritarda di diverse ore la somministrazione del farmaco l'interazione è ugualmente significativa. • Questa lunga durata d'azione deriva da un'inattivazione intestinale del CYP 3 A 4 di tipo "suicida", intendendo con questo termine che il ripristino dell'attività del CYP 3 A 4 dipende dalla sintesi di nuovo enzima. • La rilevanza dell'interazione è estremamente variabile tra gli individui; in alcuni casi le concentrazioni plasmatiche di felodipina rimangono inalterate e in altri si arriva a livelli 8 volte superiori rispetto ai controlli (felodipina + acqua). Ciò dipende dal contenuto intestinale di CYP 3 A 4: gli individui con i livelli più elevati sono quelli con i maggiori incrementi nelle concentrazioni plasmatiche di felodipina
Farmaci e succo di pompelmo Classe Farmaci Ansiolitici Buspirone, diazepam ↓ capacità psicomotorie, midazolam, triazolam↑ della sedazione Antiaritmici Amiodarone Antidepressivi Clomipramina Antiepilettici Carbamazepina Antistaminici Terfenadina Possibili eventi avversi Aritmie Sonnolenza, depressione resp. Sonnolenza, atassia, nausea Amlodipina, felodipina Calcioantagonisti Aritmie, prolungamento Nifedipina, nimodipina Q-T
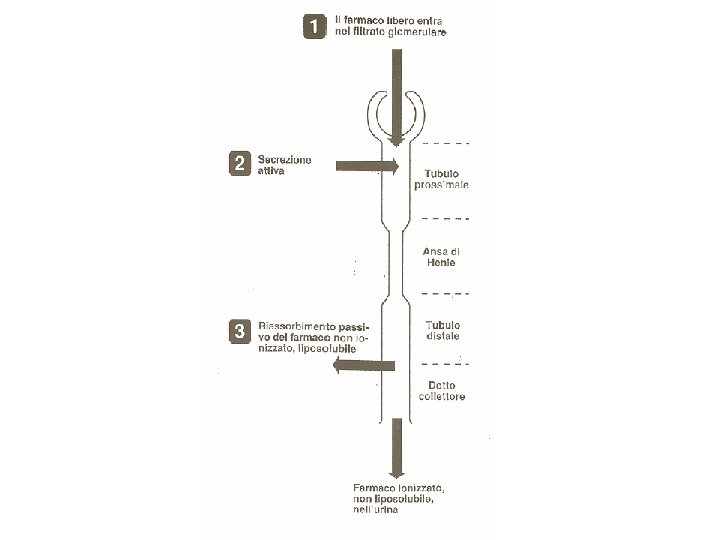
ELIMINAZIONE DEI FARMACI Ø La principale via di eliminazione dei farmaci (e dei loro metaboliti) è il RENE Ø Altre vie di eliminazione possono essere la VIA RESPIRATORIA, ad esempio per i farmaci gassosi o la VIA BILIARE (escrezione con le feci) Ø I farmaci che vengono eliminati tramite la bile possono venire in parte riassorbiti a livello intestinale: si viene cosi a creare il cosidetto CIRCOLO ENTERO-EPATICO Ø Altra via di eliminazione dei farmaci è rappresentata dal LATTE MATERNO. Questo fatto deve essere tenuto in considerazione quando si prescrivono farmaci a madri che allattano, per i possibili rischi di tossicità a cui si può esporre il neonato Ø Fattori che possono modificare l’eliminazione dei farmaci, con possibile ACCUMULO, sono: presenza di patologie renali (insufficienza renale), l’età del paziente (neonati e anziani), ostacolo al deflusso biliare (per farmaci eliminati per questa via).
Variazioni della clearance renale La capacità di eliminazione (clearance) del rene varia con l’età. E’ bassissima alla nascita ma è poi molto alta nella prima infanzia fino a scendere gradualmente nell’età adulta. (vedi figura)
ELIMINAZIONE DEI FARMACI Nella grande maggioranza dei casi, l’eliminazione dei farmaci dal corpo segue una cinetica monoesponenziale con base e. Questo significa che viene eliminata nell’unità di tempo una percentuale fissa del farmaco presente nel corpo Quindi, se un certo farmaco è eliminato al 10% all’ora e la sua concentrazione è di 10 mg/l alle ore 15. 00, alle ore 16. 00 sarà 9 mg/l; alle 17. 00 sarà 8, 1 mg/l; alle 18. 00 sarà 7, 3 mg/l; alle 19. 00 sarà 6, 6 ecc Quando una curva monoesponenziale viene disegnata su un grafico semilogaritmico (asse x lineare; asse y logaritmico in cui ogni tacca della stessa lunghezza corrisponde ad un raddoppio del valore) essa diventa una retta Il dato cinetico più informativo di una cinetica monoesponenziale è l’emivita L’emivita di un farmaco è il tempo necessario perché la concentrazione diventi la metà di quella che era al primo punto
ELIMINAZIONE DEI FARMACI Poiché la riduzione della concentrazione in scala semilogaritmica è una retta, il valore di emivita è sempre lo stesso, indipendentemente dal momento in cui si considera il primo punto (vedi figura sottostante)
ELIMINAZIONE DEI FARMACI L’insufficienza renale può modificare sensibilmente i processi di eliminazione dei farmaci. Negli esempi sottostanti si può vedere come aumenta l’emivita di eliminazione (espressa in ore) di alcuni farmaci in presenza di anuria.
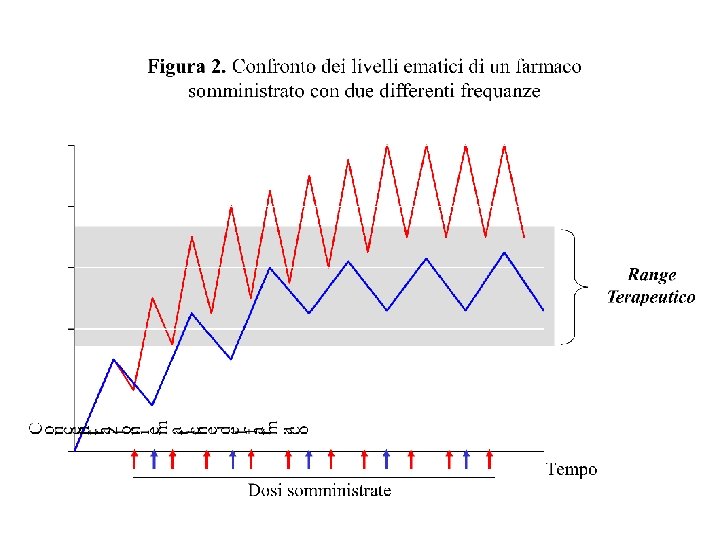
EMIVITA L’emivita permette di stimare quanto tempo deve passare perchè buona parte del farmaci sia eliminata. Per un farmaco con emivita di 1 ora, la sua concentrazione diventa il 50% dopo un’ora, il 25% dopo due ore; il 12, 5 dopo 3 ore, il 6, 25 dopo 4 ore e il 3, 12 dopo 5 ore. Se il farmaco ha emivita di 8 ore, la sua concentrazione si sarà ridotto al 3% 40 ore (5 emivite moltiplicato 8 ore) Somministrando un farmaco a intervalli di una emivita si ottengono minime oscillazione della concentrazione ematica intorno a quella terapeutica (steady-state) Sono necessarie circa 5 -7 emivite per raggiungere lo steady-state Sono necessarie almeno 7 emivite per ottenere il wash-out del farmaco
DURATA D’AZIONE DEI FARMACI Ø I farmaci possono avere differenti durate d’azione (si parla di farmaci ad azione breve, talora ultrabreve, intermedia, lunga). La durata d’azione di un farmaco dipende principalmente: o Dalla velocità di eliminazione o Dai processi di biotrasformazione (metaboliti inattivi o attivi) Ø La velocità di eliminazione dipende dalla funzionalità degli organi emuntori, dalle caratteristiche chimico-fisiche del farmaco o dei metaboliti (in particolare l’idrosolubilità), dalla forma farmaceutica, dalla via di introduzione. Normalmente la dose non influenza la velocità di eliminazione tranne che non si somministrino dosi molto elevate, tali da saturare i processi di eliminazione.
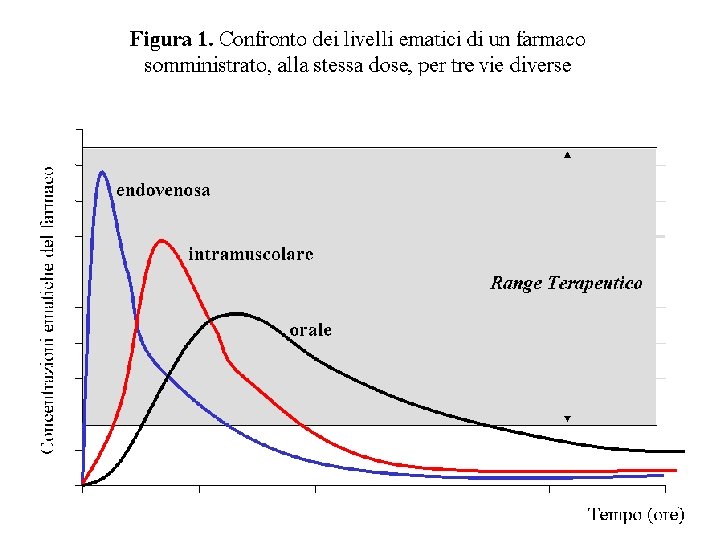
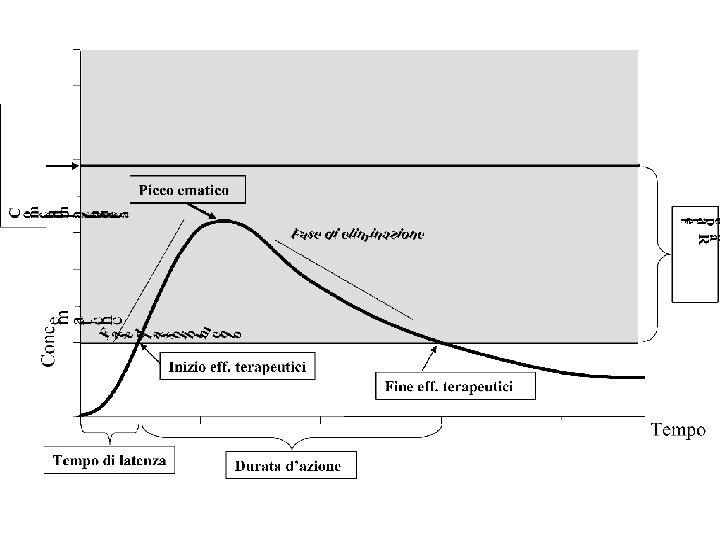
LIVELLI EMATICI (CONCENTRAZIONI EMATICHE) DEI FARMACI Ø Rappresentano la quantità di farmaco attivo contenuta nel sangue nel tempo. Ø Esiste una corrispondenza tra i livelli ematici di un farmaco e la quantità di farmaco che raggiunge la sede d’azione. In altre parole vi è corrispondenza tra i livelli ematici e l’effetto farmacologico. Ad esempio il massimo effetto di un farmaco si avrà nel momento in cui è massima la concentrazione del farmaco nel sangue.
LIVELLI EMATICI (CONCENTRAZIONI EMATICHE) DEI FARMACI Ø I livelli ematici di un farmaco dipendono da diversi fattori, quali: § la via di somministrazione § la quantità e velocità dell’assorbimento § la velocità di eliminazione § la modalità di somministrazione (unica o ripetuta, nel secondo caso ha rilievo l’intervallo di tempo tra le somministrazioni) § la quantità di farmaco somministrata (DOSE)
RANGE TERAPEUTICO L’intervallo di concentrazioni ematiche di un farmaco entro il quale si manifestano normalmente gli effetti terapeutici senza effetti tossici dosedipendenti CONCENTRAZIONE MINIMA TOSSICA La concentrazione ematica di un farmaco al di sopra della quale compaiono gli effetti tossici dose-dipendenti. Corrisponde al limite superiore del range terapeutico CONCENTRAZIONE MINIMA TERAPEUTICA La concentrazione ematica di un farmaco al di sotto della quale non si hanno effetti terapeutici. Corrisponde al limite inferiore del range terapeutico.
PICCO EMATICO La concentrazione massima raggiunta da un farmaco. Si correla al tempo. Ad esempio il picco ematico dell’aspirina somministrata per via orale si ottiene, generalmente, dopo 2 ore dalla somministrazione EMIVITA (T½) Il tempo necessario perché la concentrazione ematica di un farmaco diventi la metà. Normalmente si esprime in ore TEMPO DI LATENZA Il tempo necessario, dopo la somministrazione, per ottenere l’inizio dell’effetto del farmaco. Quindi il tempo necessario ad ottenere la minima concentrazione terapeutica
FINE DELL’EFFETTO TERAPEUTICO Il tempo trascorso dalla somministrazione alla fine dell’effetto del farmaco. Quindi il tempo per raggiungere nuovamente una concentrazione ematica al di sotto di quella minima terapeutica DURATA D’AZIONE L’intervallo di tempo tra l’inizio e la fine degli effetti terapeutici di un farmaco. Quindi il tempo in cui i livelli ematici sono all’interno del range terapeutico
INDICE TERAPEUTICO Ø L’indice terapeutico di un farmaco è rappresentato dal numero derivante dal rapporto tra la dose tossica e la dose terapeutica. Ø Ad esempio per un farmaco che ha una dose tossica di 10 grammi ed una dose terapeutica di 2 grammi: Ø Risulta evidente che quanto più l’indice terapeutico di un farmaco è basso (vicino all’unità) tanto più ristretto è il margine di sicurezza nel dosaggio del farmaco.
INDICE TERAPEUTICO Ø L’indice terapeutico non rappresenta la valutazione di un farmaco dal punto di vista dell’efficacia e/o della tollerabilità ma ci indica soltanto la vicinanza o meno della dose tossica rispetto a quella terapeutica. Ø Farmaci con un basso indice terapeutico (ad esempio antiepilettici, teofillina, aminoglicosidi, antitumorali, warfarin) devono essere monitorati. Il monitoraggio si può effettuare direttamente, cioè prelevando dei campioni di sangue e determinando la quantità di farmaco presente, o indirettamente attraverso dei parametri di laboratorio, ad esempio per il warfarin o altri anticoagulanti misurando il tempo di coagulazione del sangue. In base ai risultati ottenuti si aggiusta la dose da somministrare.
RAPPORTO RISCHIO/BENEFICIO Ø La valutazione clinica di un farmaco è un processo complesso non esprimibile con un semplice rapporto tra dose tossica e dose terapeutica (indice terapeutico). Si tratta, infatti, di esprimere un giudizio valutando da una parte i benefici che si ottengono e dall’altra i rischi che si corrono utilizzando il farmaco (rapporto beneficio/rischio). Ø Per stabilire un corretto rapporto beneficio/rischio per un farmaco è necessario conoscere i benefici che si ottengono (quindi conoscere i dati sulla sua efficacia clinica) e i rischi derivanti dal suo uso (quindi conoscere i suoi effetti avversi). Ø Bisogna tenere presente che il rapporto beneficio/rischio di un farmaco può essere diverso a seconda del paziente e/o della patologia da trattare. Quindi in certe situazioni un farmaco, che ha in generale un rapporto beneficio/rischio favorevole (cioè i benefici superano i rischi), potrebbe avere un rapporto sfavorevole (i rischi superano i benefici).
DOSAGGIO: DEFINIZIONI DOSE Quantità di farmaco somministrata per produrre un determinato effetto terapeutico POSOLOGIA Dose, tempi e modalità di somministrazione di un farmaco Esempio di posologia: Rocefin 500 mg due volte al giorno per via i. m. per 7 giorni DOSE MASSIMA La massima quantità di farmaco tollerata, senza cioè che si verifichino effetti tossici DOSE GIORNALIERA La quantità di farmaco somministrata nelle 24 ore
FATTORI DA CONSIDERARE NELLA DETERMINAZIONE DELLA DOSE Ø Modalità di somministrazione Ø Peso (per farmaci ad alto rischio con basso indice terapeutico meglio utilizzare la superficie corporea) Ø Età Ø Patologie concomitanti Ø Gravidanza Ø Contemporanea somministrazione con altri farmaci che interagiscono
PRINCIPALI UNITA’ DI MISURA DEI FARMACI Ø Microgrammo (mg o mcg) o gamma (g): millesima parte del milligrammo Ø Milligrammo (mg): millesima parte del grammo Ø Grammo (g) Esempi: 400 mcg = 0, 4 mg; 2 mg = 2000 mcg; 500 mg = 0, 5 g; 3 g = 3000 mg Ø Unità internazionali (UI): quantità di farmaco che provoca un determinato effetto biologico [esempi di farmaci per cui si utilizzano le UI: insulina, eparina, eritropoietina, fattori della coagulazione, penicillina G, interferone, immunoglobuline, calcitonina]
MISURE DI CAPACITA’ Ø Microlitro (ml): millesima parte del millilitro Ø Millilitro (ml): millesima parte del litro Ø Centilitro (cl): centesima parte del litro Ø Decilitro (dl): decima parte del litro Ø Litro (L) Esempi: 400 ml = 0, 4 ml; 2 ml = 2000 ml; 500 ml = 0, 5 L; 50 cl = 0, 5 L; 10 dl = 1 L
SOLUZIONI DEI FARMACI Ø Soluzione al 5% = 5 grammi di farmaco in 100 ml Ø Soluzione al 2% = 2 grammi di farmaco in 100 ml Ø Soluzione al 9 per mille = 9 grammi in 1000 ml (1 L) questi sono esempi di concentrazioni peso/volume, nel caso di pomate o unguenti le concentrazioni sono peso/peso ad esempio pomata al 5% = 5 grammi di farmaco in 100 g di pomata