Enfermedades musculares Dr Miguel A Barboza Elizondo Departamento

Enfermedades musculares Dr. Miguel A. Barboza Elizondo Departamento de Neurología Hospital Rafael A. Calderón Guardia
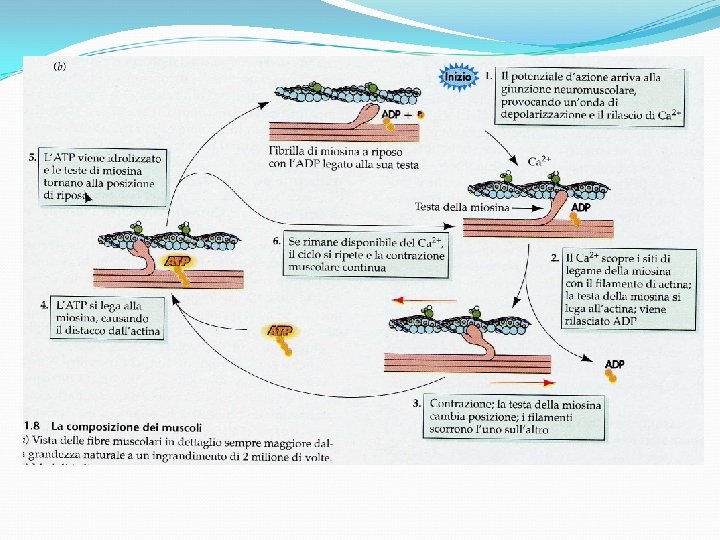


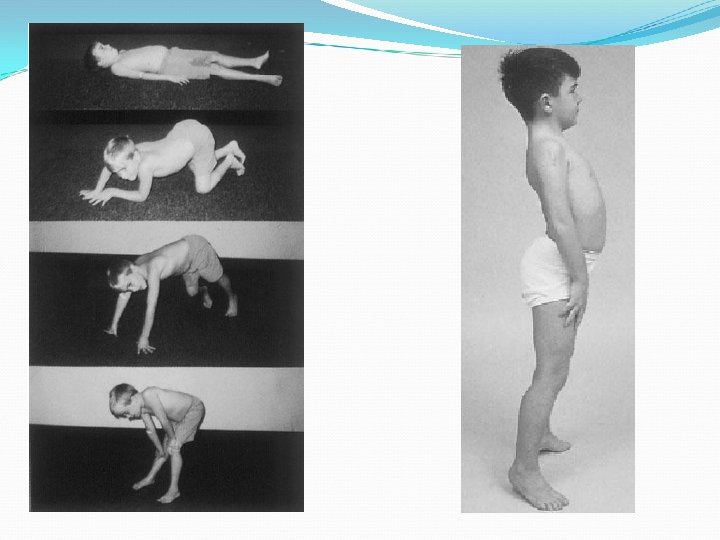
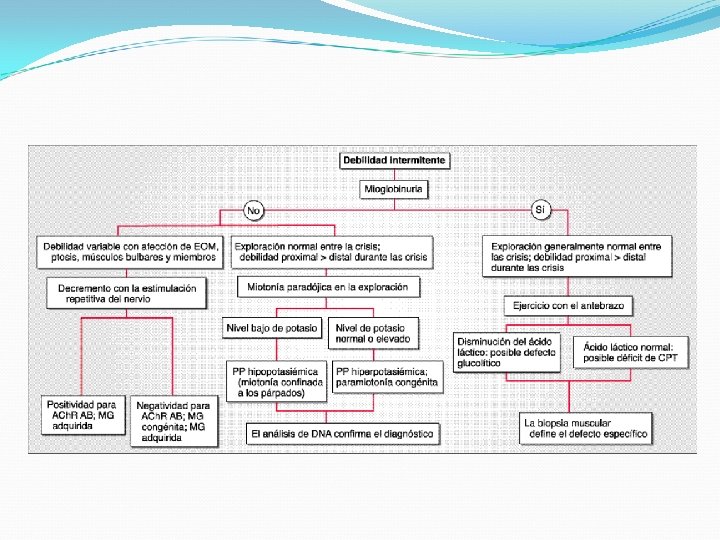
Características clínicas de enfermedades musculares Debilidad intermitente: Miastenia Gravis, Parálisis periódica, déficit energéticos -metabólicos (glicólisis y ac. Grasos) Debilidad persistente: Distrofia muscular, polimiositis, dermatomiositis Debilidad predominantemente proximal, ocasionalmente puede incluir músculos PC Variación clínica de acuerdo a etiología Signo de Gowers


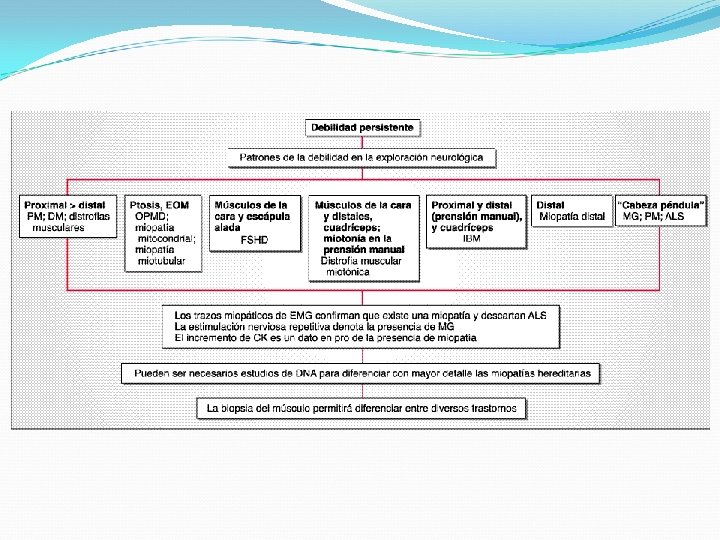
Características clínicas Fatiga: imposibilidad de mantener la fuerza Mialgias pueden ser localizadas o generalizada, y no necesariamente se acompaña de debilidad Espasmo muscular (calambre): contracción muscular localizada, involuntaria y dolorosa repentina Contractura muscular: endurecimiento muscular por un fracaso energético en los trastornos de la glicólisis Miotonía: contracción muscular prolongada con relajación lenta Hipertrofia/atrofia

Distrofias musculares Grupo de entidades hereditarias progresivas cuyo denominador común resulta en la afectación muscular Patrones fenotípicos y genotípicos identificadores Frecuencia baja en población general, con variación por grupos raciales y zonas geográficas

Distrofia muscular de Duchenne Recesiva ligada al cromosoma X 30 por 100000 varones nacidos vivos Presente al nacer con manifestaciones entre 3 -4 años Evidente debilidad a los 5 años 6 años: contracturas del tendón de Aquiles y las bandas iliotibiales + posturas lordóticas Progresión a debilidad proximal y flexores del cuello 8 -10 años: muletas para deambulación 12 años: silla de ruedas + escoliosis restrictiva

Distrofia muscular de Duchenne Alteración progresiva de la función pulmonar Infecciones pulmonares Broncoaspiración Gran porcentaje de miocardiopatía Deterioro intelectual progesivo CPK elevada entre 20 y 100 veces Etiología: mutación de gen que codifica distrofina Alteración de los complejos estables que se forman con los sarcoglucanos, con deficiencia de otras proteínas estructurales
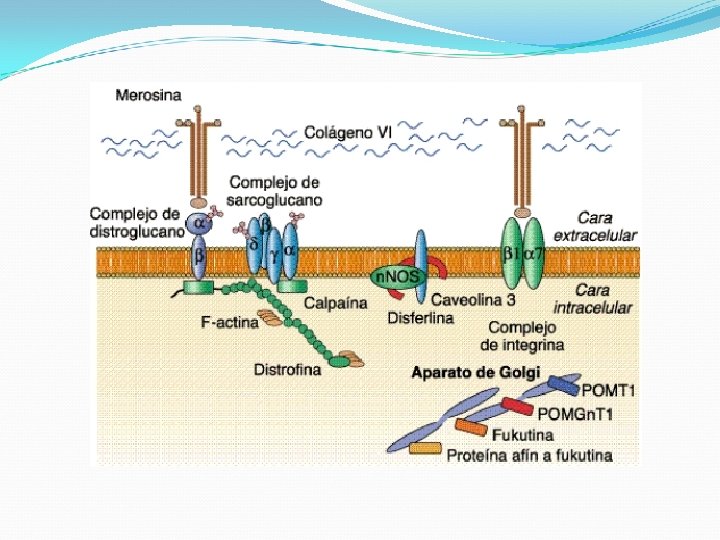

Distrofia muscular de Becker Ligada al cromosoma X 10 veces menos frecuente que Duchenne (3/100000) Manifestaciones clínicas: Afectación proximal músculos Ms. Is>Ss Hipertrofia precoz de pantorrillas Inicio de síntomas entre 5 -15 años (aún a los 15 pueden caminar) Sobrevida a 4 -5° década Retraso mental menos frecuente Afección cardíaca usual
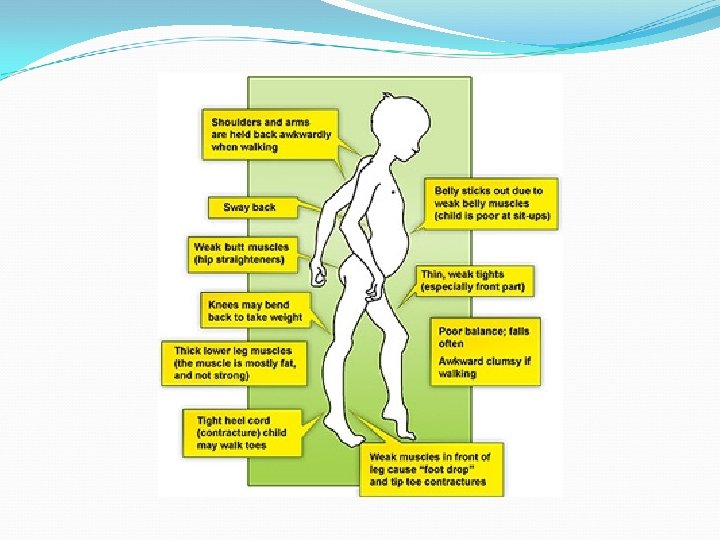
 Afección de diversos grados de debilidad")
Distrofia muscular congénita No una entidad única (AR) Afección de diversos grados de debilidad muscular, alteración del SNC y anomalías oculares Se presentan desde el nacimiento o los primeros meses Patrón miopático debilidad que puede incluir músculos faciales, pero respeta extraoculares Artrogriposis: contracturas al nacimiento Afectación cerebral severa: Distrofia muscular congénita de Fukuyama, enfermedad músculo-ojocerebro y el síndrome Walker-Warburg Alteración Visual: Fukuyama
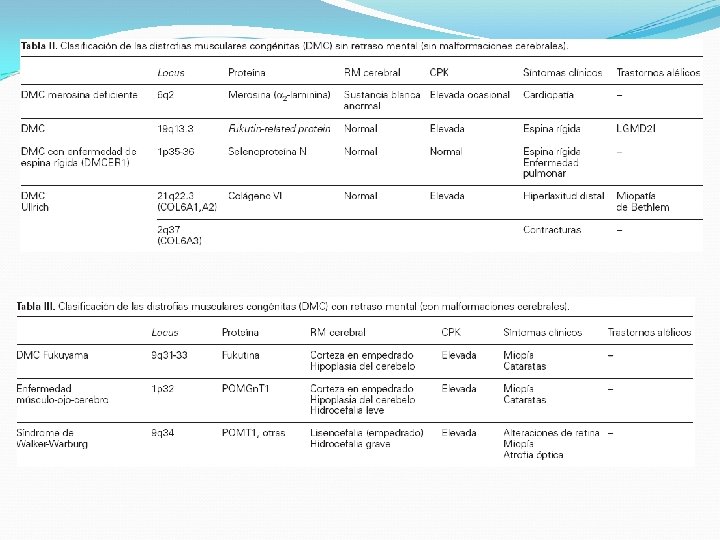

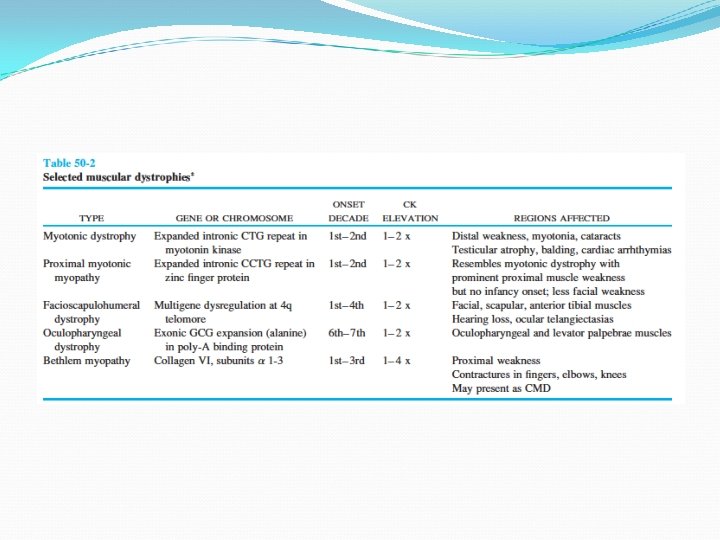
Distrofia miotónica DM tipo 1: Steinert y DM tipo 2: Miopatía miotónica proximal Clínica “cara de hacha” Afectación de músculos del cuello Afectación de músculos distales de los miembros Déficit extensor de muñecas y pie péndulo Puede haber disartria por disfunción palatina, faríngea y lingual + voz nasal + disfagia miotonía

Distrofia miotónica DM tipo 1 Trastornos del ritmo cardiaco, déficit intelectual, hipersomnio, cataratas subcapsulares posteriores, atrofia gonadal, resistencia a la insulina DM tipo 2 Más grave Debilidada facial y bulbar intensa que puede llevar a insuficiencia respiratoria neonatal y retardo mental Afección de predominio proximal Comparte algunas características de la DM tipo 1

Dermatomiositis y polimiositis

Definición Enfermedad difusa e inflamatoria de origen autoinmune que afecta al músculo estriado asociado a síntomas cutáneos. Puede ser aguda, subaguda o crónica Afecta principalmente músculos proximales

Dermatomiositis • Con síntomas cutáneos Polimiositis • Sin síntomas cutáneos

Epidemiología 1 -6 casos/ 1000 habitantes Más frecuente en mujeres Afroamericanos 5 -15 años de edad 5 ta década de vida

Etiología Desconocida Autoinmune Factores ambientales, infecciosos y genéticos Neoplasias

Etiopatogenia Músculo estriado Citotoxicidad mediada por linfocitos T Complemento de ataque de membrana Partículas virales HLA-88 HLA B 14 genético Respuesta inmunológica anormal en neoplasias Distinto en niños
 2. Tipo II: polimiositis y")
Tipos 1. Tipo 1: miositis sin lesiones cutáneas (polimiositis) 2. Tipo II: polimiositis y lesiones cutáneas (dermatomiositis clásica) 3. Tipo III: polimiositis o dermatomiositis acompañada de neoplasia (paraneoplásica) 4. Tipo IV: polimiositis o dermatomiositis infantil o juvenil (lenta o benigna) 5. Tipo V: polimiositis o dermatomiositis relacionada con otra enfermedad del tejido conectivo. 6. Tipo VI: dermatomiositis sin miositis (amiopática)

Dermatomiositis Juvenil Edad media de aparición es de 9 años No existe asociación con tumores

Forma paraneoplásica 15 a 20% de los casos Después de los 40 años de edad Predominante en los varones.

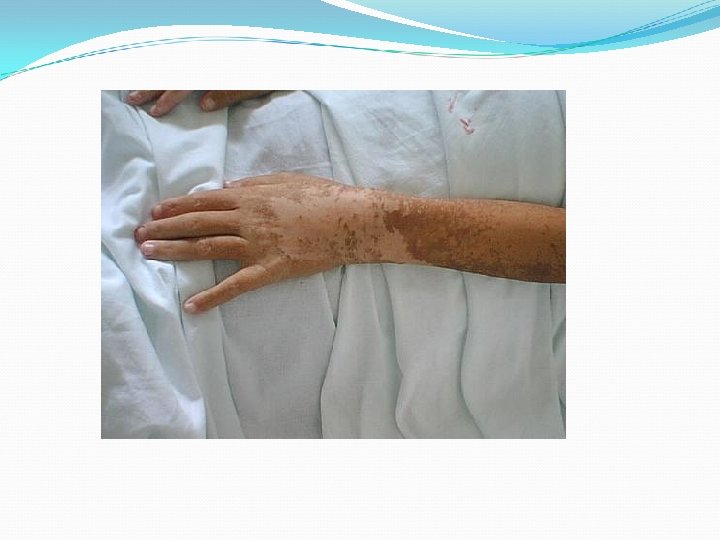
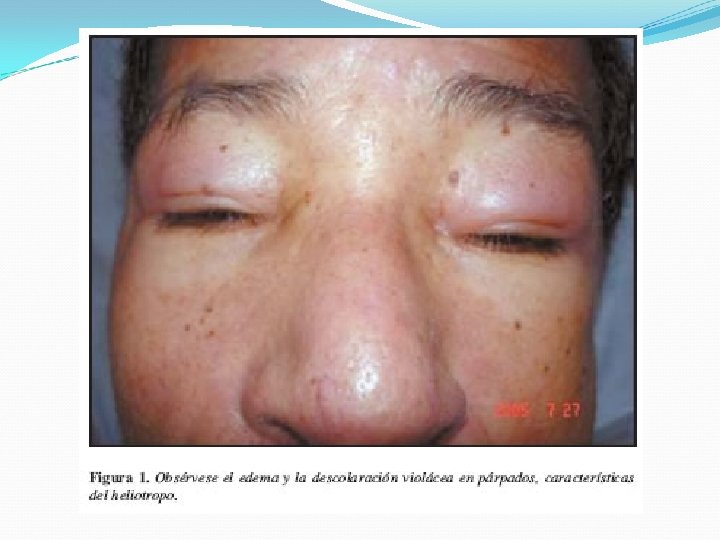
Clínica Inicio puede ser repentino o insidioso Inicialmente se caracteriza por astenia y debilidad muscular síntomas cutáneos y generales Evolución es crónica y lentamente incapacitante. Síntomas cutáneos: párpados, frente, regiones malares y dorso de la nariz Eritema de color lila, descamación y
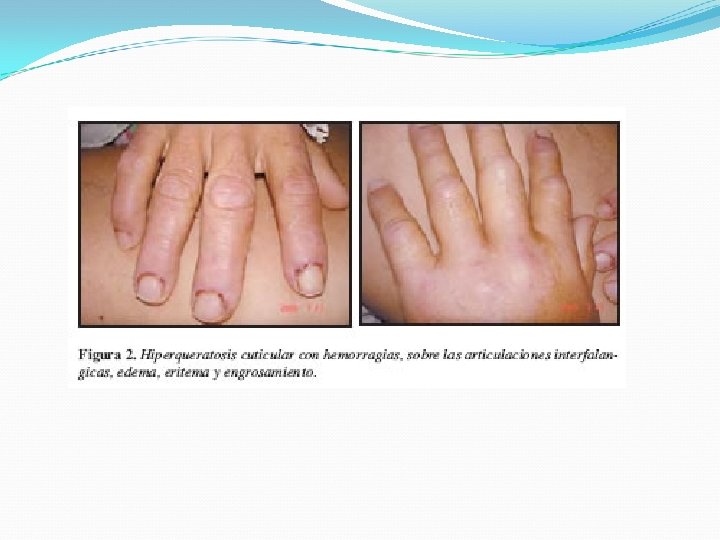

Lesiones de aspecto poiquilodérmico : eritema, atrofia, pigmentación y telangiectasias Signo de Gottron 1/3 Pápulas violáceas planas que desarrollan atrofia, telangiectasias e hipopigmentación sobre las superficies articulares de los dedos. Fenómeno de Raynaud Vasculitis Telangectasias periungueales
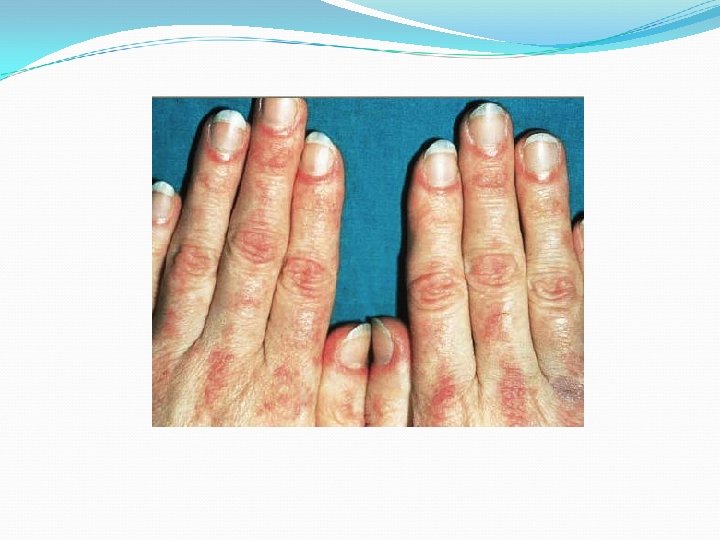

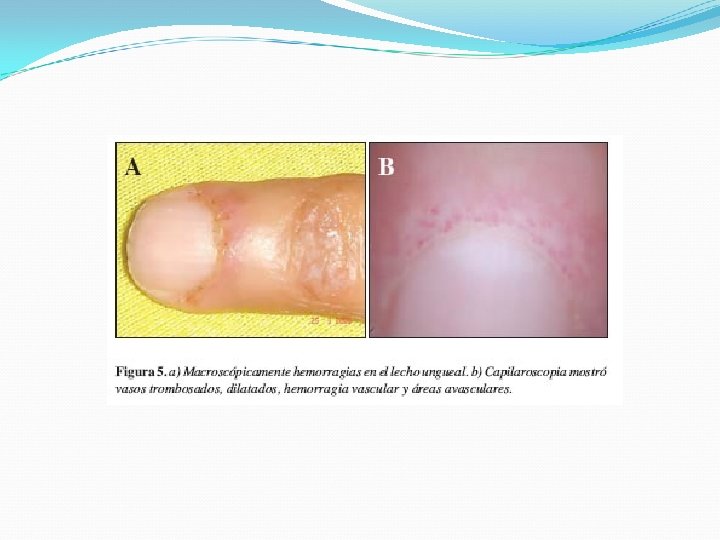

Manifestaciones musculares Debilidad simétrica de la musculatura esquelética Predominio proximal (hombro y cintura pélvica femoral) Afectación de musculatura deglutoria, respiratoria y cardiaca Acompañado de síntomas sistémicos como astenia, pérdida de peso, febrícula, anorexia, edema y artralgias.

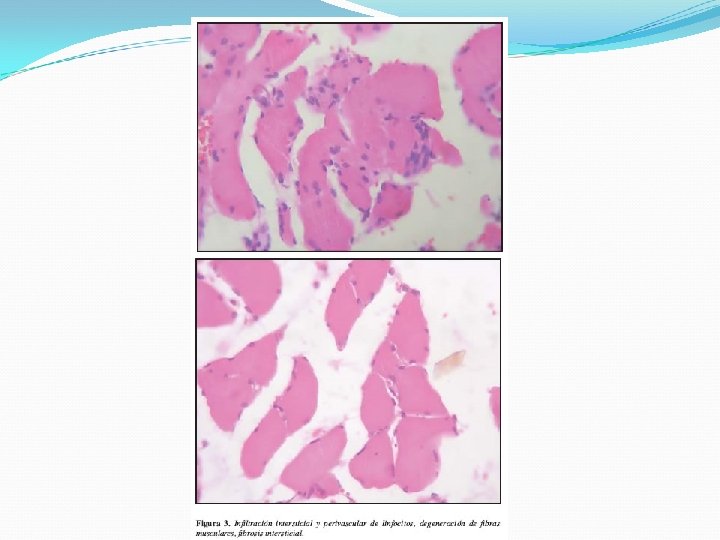
Criterios diagnósticos 1. Debilidad muscular proximal simétrica. 2. Biopsia muscular anormal. 3. Aumento de enzimas séricas músculo específicas. 4. Electromiografía anormal. 5. Erupción cutánea típica La presencia de la erupción típica proporciona certeza diagnóstica; con tres o cuatro de los restantes es definitivo; con dos, probable, y con uno, posible.

La mortalidad ronda el 25%; puede controlarse con el tratamiento adecuado y se puede tener una sobrevivencia del 85%.

Diagnóstico Dosis elevadas de prednisona Metotrexate Globulinas Evaluación de causas subyacentes

En conclusión…. La dermatomiositis es una enfermedad inflamatoria autoinmune que afecta predominantemente músculos proximales y puede o no tener manifestaciones cutáneas. Existen diferentes tipos de esta enfermedad, siendo los principales la dermatomiositis juvenil, paraneoplásica y dermatomiositis clásica. La clínica presenta un amplio espectro, desde manifestaciones levas hasta la muerte principalmente debido a afectación de musculatura

¡Muchas Gracias!
- Slides: 45