Efnaskiptasjkdmar og taugakerfi Sigurur Marelsson Barnalknir Helstu flokkar

Efnaskiptasjúkdómar og taugakerfið Sigurður Marelsson Barnalæknir
 • Hvaða")
• Helstu flokkar efnaskiptasjúkdóma sem gefa einkenni frá taugakerfinu (neurometabolic disease) • Hvaða einkenni • Hvaða rannsóknir eru gerðar við uppvinnslu þessara sjúkdóma • Afhverju er mikilvægt að greina?

Neurometabolic disease • Lysosomal disease • Mitochondrial disease • Peroxisomal disease
 (Wilson disease, Menkes disease) • Vítamínum")
Sjúkdómar tengdir • Málmum (Disorders of Metal Metabolism) (Wilson disease, Menkes disease) • Vítamínum (Vitamin/Cofactor deficiencies) (Pyridoxine or Biotinidase deficiency) • Flutningi efna (Disorders of Transport) (Creatine transporter deficiency, Glut-1 transport defect) • Púrín/pýramídin (Disorder of Purines/pyrimidines) (Lesch Nyhan Disease) • Óeðlileg ummyndun amínósýra (Amino Acidurias) (Maple syrup urine disease, nonketotic hyperglycinemia) • Óeðlilegri lífrænni sýrumyndun (Organic acidurias) (Methylmalonic acidemia, glutaric aciduria type 1) • Disorders of Urea Cycle
")
Sjúkdómar tengdir • Disorders of Carbohydrate metabolism (Glycogen storage disease, congenital disorder of glycosylation) • Beta fitusýru-oxunargallar (Disorders of Fatty Acid Oxidation)
 td. methylmalonic acidemia (propionic acidemia, multiple caroxylase")
• Organic acidemias (óeðlileg lífræn sýrumyndun) td. methylmalonic acidemia (propionic acidemia, multiple caroxylase defeciency) vegna truflana í efnaskiptum próteina, fitu og sykra og einkennist af hækkuðu laktat og vægri hækkun NH 4. Algeng einkenni eru einkenni frá heila (encephalopathy), neutropenia og lækkun blóðflagna. Einkenni aukinnar lífrænnar sýrumyndunar koma oft fram sem lífshættulegt ástand snemma á ævinni með metabólískri acidósu, auknu anjónabili og einkennum frá taugum og líffærum. • Aminoacidopathies (óeðlileg ummyndun amínósýra )(PKU, MSUD: maple syrup urine disease, nonketotic hyperglycinemia), MSUD kemur fram í lok fyrstu viku ævi með flogum, meðvitundarskerðingu, næringarvanda og sérstakri lykt. • Nonketotic hyperglycinemia einkennist af flogum og hypotoniu. Með raðmassagreininum má mæla nær allar amínósýrur út blóðþerripappírsýninu. • Urea cycle defects (citrullinemia, arginosuccinic aciduria) einkennist af hækkun NH 4 og resp. alkalosis. • Primary lactic acidosis • Fatty ocid oxidation defects (Beta-oxidation defects). Beta fitusýru-oxunargallar. Hypoketotic hypoglycemia, hækkun á NH 4 og cardiomyopathy. Einnig hækkun acylkarnitin sem er hægt að mæla, MCAD (medium chain acyl-Coa dehydrogenase deficiency er líklegast algengast etv orsakar um 5% SIDS tilfella.
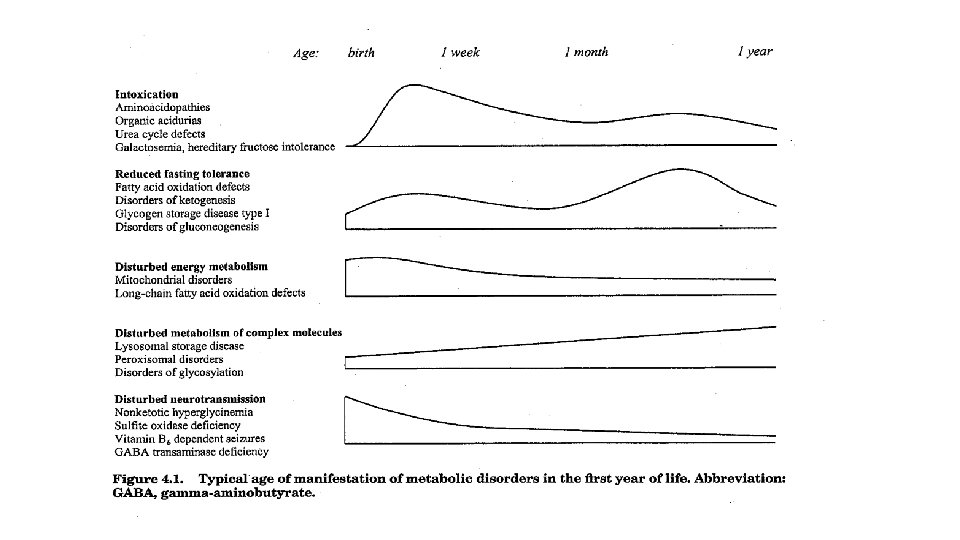

Disorders that present with acute lifethreatening illness • Disorders of fatty acid oxidation (coma, hypoglycemia, seizures) • Organic and amino acidurias (acidosis) • Mitochondrial disorders (acidosis) • Disorders of urea cycle (hyperammonemia)
 eru organic acidurias")
• Algengustu efnaskiptasjúkdómarnir sem gefa einkenni frá miðtaugakerfinu (neurometabolic disorders) eru organic acidurias og amino acid apathies en þar á eftir eru neuronal ceroid lipofuscinoses, urea cycle disorders, congenital lactic acidosis og peroxisomal disorders. • Sjaldgæfari eru sphingolipidoses, mucopolysaccharidoses, glycoprotein degradation disorders and fatty acid oxidation disorders.

Einkenni • Miðtaugakerfi • Vöðvar • Hjarta • Augu • Eyru • Meltingarvegur • Nýru • Önnur líffæri
 -hluti af almennum sjúkdómi -metabolísk krísa,")
Einkenni • Bráð einkenni frá heila (acute encephalopathy) -hluti af almennum sjúkdómi -metabolísk krísa, organiskar aciduriur, sjúkdómar í hvatberum, urea cycle defect. td. stroke, flog, meðvitundarskerðing, delerium, lífshættulegt.

Einkenni • Chronic encephalopathy • • • Þroskaskerðing, • • • Önnur einkenni Flogaveiki Hreyfiskerðing (ataxia, spasticitet) Hegðunarerfiðleikar, geðræn einkenni Heyrnarskerðing Sjónskerðing Vaxtarskerðing Lifrarstækkun Metabolic acidosis Einkenni frá meltingarvegi, uppköst
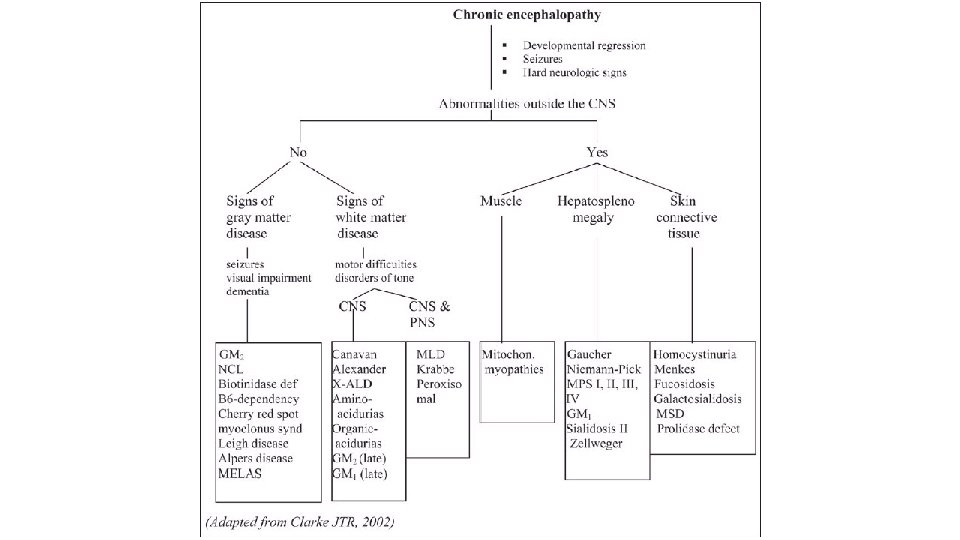

Skoðun • Almenn skoðun • Þarf að hafa í huga: • lykt • almennt útlit • bein • hár • húð • augu • Taugaskoðun

MPS
 • Óregluleg lögun beina (MPS) • Sérkennilegt útlit")
Almenn einkenni • Grófir andlitsdrættir (MPS) • Óregluleg lögun beina (MPS) • Sérkennilegt útlit (Smith Lemli Opitz) • Lágvaxinn (Mito, MPS) • Óeðlilegur hárvöxtur (hypertrichosis) (Mito) • Stórt höfuð (macrocephaly)(glutaric aciduria I, Canavan, Alexander) • Lítið höfuð (Mito)
 • Skallablettir (alopecia) (Biotin deficiency) • Angiokeratoma")
Hár og húð • Húðlitur, fölvi (PKU) • Skallablettir (alopecia) (Biotin deficiency) • Angiokeratoma (Fabrys) • Brittle hair (Menkes) • Innvaxnar geirvörtur (CDG)

Angiokeratoma

Brittle hair in Menkes disease
 (MPS) • Cataract • Cherry red spot")
Augnskoðun • Skýmyndun á augasteini (corneal clouding) (MPS) • Cataract • Cherry red spot (GM I; GM II, gangliosidosis) • Rýrnun sjóntaugar (optic atrophy) (Mito) • Litun sjónhimnu (pigmentary retinopathy) (NCL, Mito, peroxisomal) • Ptosis (NP gerð C, Mito)

Cherry red spot

Retinitis pigmentosa
 • Nýrnasjúkdómar (Fabrys,")
Lifur, milta og nýru • Lifrar og miltisstækkun (Lysosomal storage disease) • Nýrnasjúkdómar (Fabrys, Mito)

Lykt
 (Mito, peroxisomal) • Spastisitet, dystonia (Mito, organic")
Einkenni frá miðtaugakerfi • Minnkuð vöðvaspenna (hypotonia) (Mito, peroxisomal) • Spastisitet, dystonia (Mito, organic aciduria) • Flog (á fyrstu dögunum: NKH, Mito, Pyridoxin def, peroxisomal) • Peripheral neuropathy (MLD, Krabbe, Mito)

Hvenær er byrjað að leita?

• Ákjósanlegt á fyrstu 2 -5 dögum nýburans til þess að greina meðfædda efnaskiptasjúkdóma. • Einmitt á þessu tímabili þegar næring frá móðurinni fer þverrandi og nýburinn fer að fá næringu um munn og fæðuniðurbrotsferlar eru þandir til hins ýtrasta, gefast mestu möguleikarnir á að finna fitusýruoxunargalla (fatty acid oxidation disorder) og einnig galla er leiða til óeðlilegar myndunar lífrænna sýra (organic aciduria).

• Þegar eðlilegu næringarástandi er náð er möguleiki á að vísarnir verði eðlilegir að nýju fyrir suma sjúkdómana og greinist því ekki nema í “krísuástandi”. Hins vegar aukast möguleikarnir á að greina óeðlilega ummyndun amínósýra eftir að nýburinn hefur fengið næringu.

Rannsóknir • P-glukos, astrup, elektrólýtar, laktat, ketónur, NH 4, lifrarpróf, kreatinin, homocystein, B 12, Folat, methylmalonat, PTH, jóniserað Ca 2+, TSH, f. T 4. • Taka prufur fyrir: p-as, p-karnitin, p-acylkarnitin (blot-spot), lýsósomal ensím, peroxisomal sjkd( VLCF, Fytansýru, Pristansýru, plasmalogena). • Hægt að panta sérhæfðari rannsóknir (sjá blað) • Þvag: U-as, U-os. (spot þvag sent til köben) • Smásjárskoðun á blóðstroki (vacoulur í lymfocytum) • Fleiri rannsóknir: MRI, EEG, Hjartaómun, Ómun lifur, gall og bris. • Aðrar sérhæfðar rannsóknir.

Bráðveikt barn-grunur um neurometabolic disease • Astrup • Sykur • Blóðsölt • NH 4 • Laktat, pyruvat • Plasma acylcarnitines • Plasma aminoacids • acylglycines, organic acids



Peroxisómal sjúkdómar • Mynda og brjóta niður mikilvæg efni • Í peroxisómum á sér stað beta oxun langra keðja fitusýra (VLCFA) og myndun sérstakra fosfolípiða sem eru í hjarta, heila, og vöðva og myelin. Einnig myndast kólesteról og gallsýrur. • Zellwegers syndrome og adrenoleucodystrophy eru algengust peroxisómal sjúkdómarnir. • Peroxisomal screening: Mæla VLCF, Fytansýru, Pristansýru, plasmalogena.
")
Klassískur Zellweger • Einkennandi útlit • Hypotonia • Krampar • Augneinkenni (retinitis pigm. ) • Heyrnarleysi • Áhrif á lifur • Nýru • Beinbreytingar • Áhrif á myndun heila

Sjúkdómar í hvatberum • Hópur sjúkdóma sem orsakast af því að hvatberar starfa ekki rétt. • Hvatberar eru inni frumum líkamans og framleiða orku (ATP) • Þegar orkuframleiðslan er skert geta komið fram einkenni frá mismunandi líffærum, oftast frá mörgum líffærum (heila, vöðvum, lifur osv. frv. ) • Dæmi um slíka sjúkdóma eru: Alpers sjúkdómur, Leigh sjúkdómur, MELAS og MERRF. • 5 -10/100. 000 • Orsakast af stökkbreytingum í erfðaefni í hvatberum (mt-DNA) eða í kjarna (90%). • Oft hækkað laktat (60%)

Truflun í orkuframleiðslu
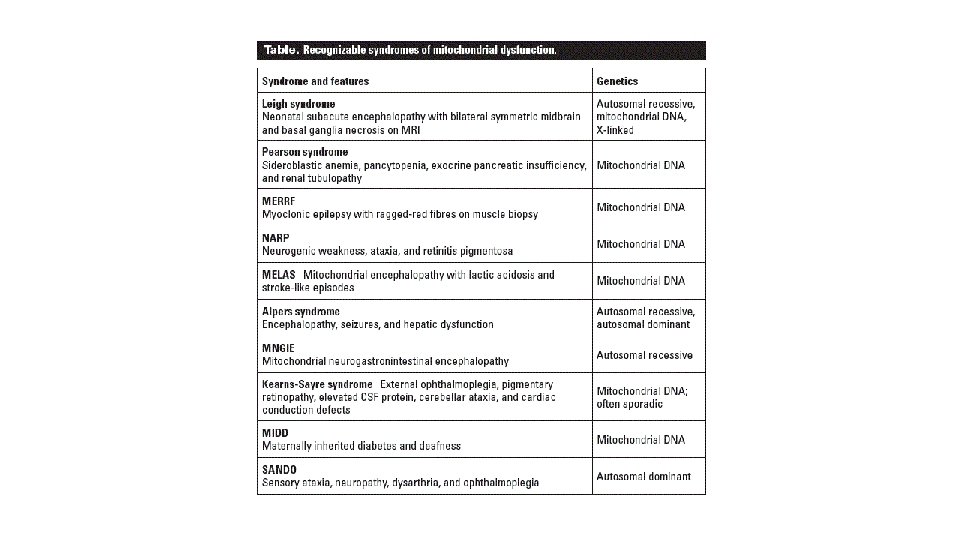

Leigh syndrome • Algengasti sjúkdómur í hvatberum • Einkenni byrja fyrir 1 árs aldur • Greining á MRI, symmetrískar breytingar í basal ganglia, heilastofni og/eða annars staðar í heila. • Versnandi neurologisk einkenni, einkenni frá basal ganglia og heilastofni.

MRI-Leigh syndrome

Greining

Lýsosómal sjúkdómar • 1/5000 nýfæddum • Vöntun á ensími • Þessi vöntun á ensími veldur upphleðslu af efnum sem ekki eru brotin niður í frumum og vefjum. • Versnandi einkenni • Mörg líffæri, dæmi um slíka sjúkdóma: Krabbes sjúkdómur, MPS, GM I, II, Pompes, margir fleiri
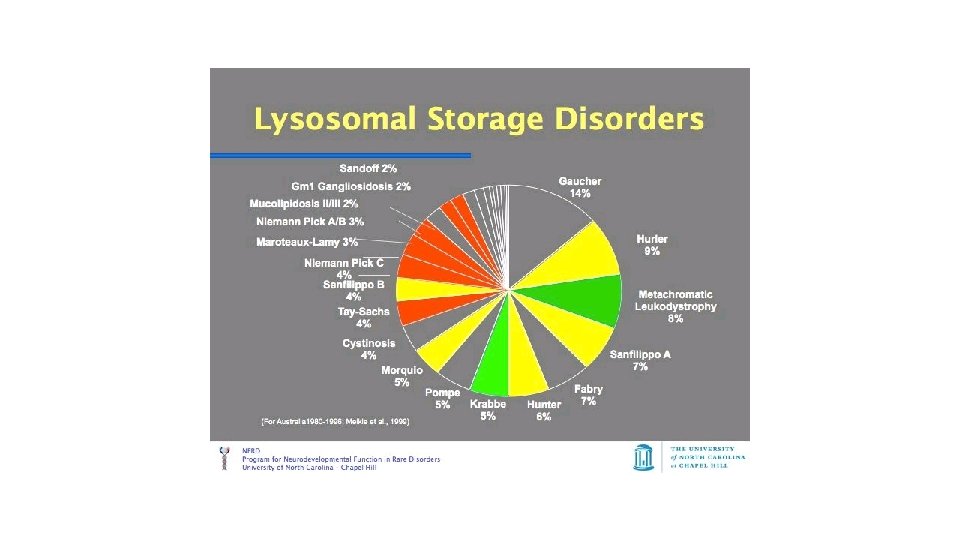

Greining • Sérstök útlitseinkenni • Líffærastækkanir • Rannsóknir • Þvagpróf • Blóðpróf í genetik • Húðsýni

MPS: Mucopolysaccharidosis • Versnandi einkenni • Einkenni frá CNS, skelett, liðum, augum, eyru, lungum, hjarta og meltingarvegi. • Gróf í andliti og stuttvaxin

MPS

Tilfelli • 6 mánaða gamall drengur fæddur eftir eðlilega meðgöngu og fæðingu. Fullburi. • Minnkuð vöðvaspenna (hypoton), veltir sér ekki, glaðlegur, nærist þokkalega, líður illa á maga, foreldrum finnst hann linur. Ungbarnaeftirlit vill bíða og sjá. • Kemur á BMT með hósta og kvef og öndunarerfiðleika. • Blásturshljóð heyrist • Leggst inn. Gerð hjartaómun, sem sýnir væga þykknun á hjartavöðva. EKG með sérkennandi útlit. Öndunarerfiðleikar svara vel meðferð. Nákvæmari skoðun er erfitt að fá fram reflexa. • Greining: Pompes sjúkdómur.
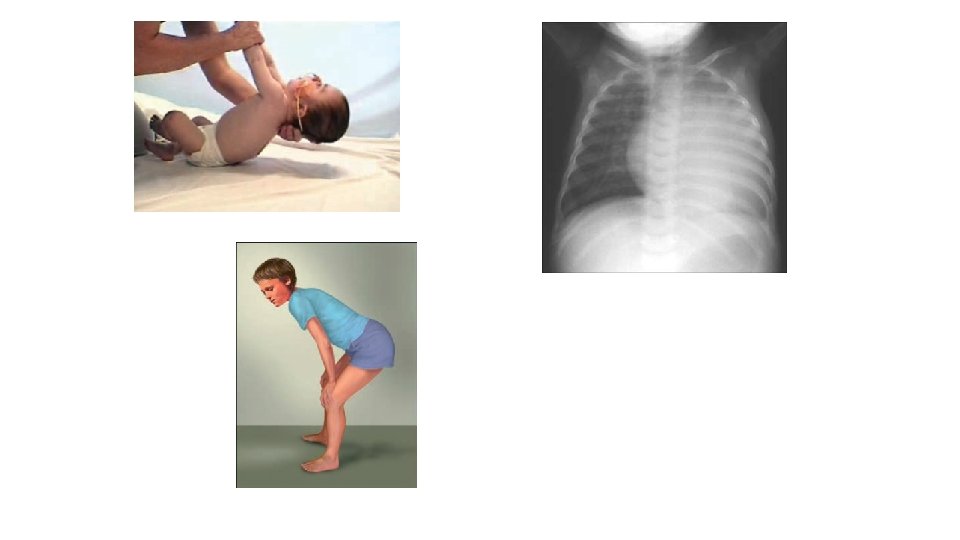
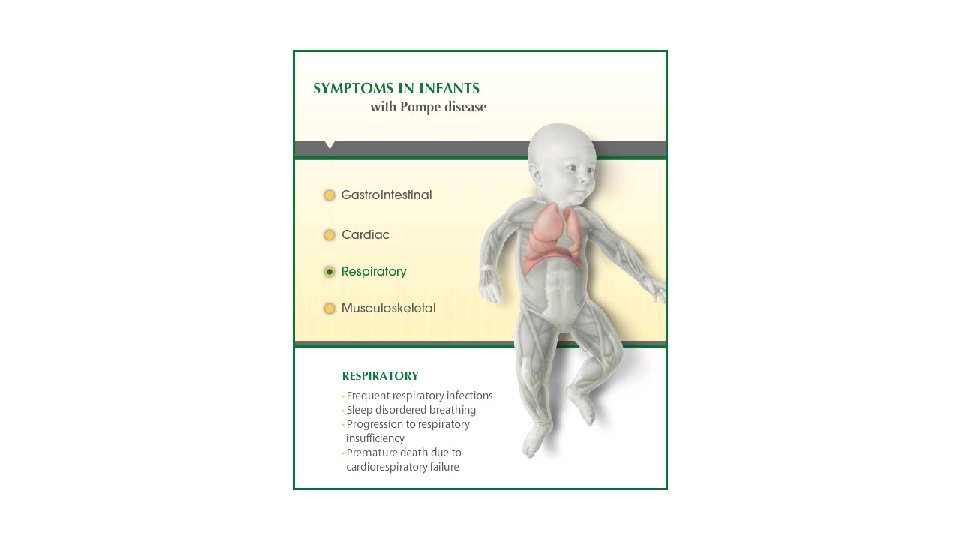

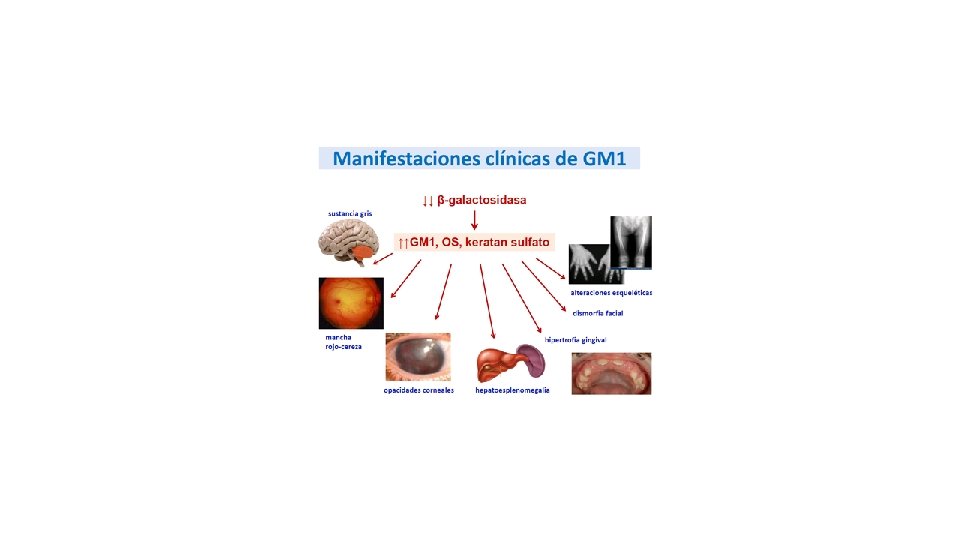
Tilfelli • 1 árs drengur, fæddur eftir eðlilega meðgöngu og fæðingu. Fullburi. • Hypoton frá fæðingu • Uppvinnsla: vacuolur í lymfocytum. • Lysosomal ensím • GMI

Wilson Disease • Truflun í kopar efnaskiptum vegna stökkbreytingar í ATP 7 B geni • Einkenni frá lifur, miðtaugakerfi, blóðmynd og geðræn einkenni • Lágt kopar og cerulloplasmin • Hækkað kopar í þvagi • Samsöfnun í lifur • Kayser-Fleischer rings
 rings")
Kayser-Fleischer (KF) rings

Neuronal Ceroid Lipofuscinosis • 1: 12. 500 • Flog, dementia, sjóntap, cerebral atrophy • juvenile neuronal ceroid lipofuscinosis (JNCL = CLN 3) or Spielmeyer. Vogt’s type • 5 -8 ára, dementia, ataxia, spastisitet, dystonia • Cerebral atrophy, retinitis pigmentosa
- Slides: 55