Drug Absorption Distribution Metabolism Elimination Chapter 3 PhysicalChemical

Drug Absorption, Distribution, Metabolism, Elimination Chapter 3

Physical/Chemical Properties of Drugs Ability to Approach Receptors
 – Bulk flow transfer – Fast, long-distance")
Drug Mol’s Receptors • Bloodstream (cardiovascular system) – Bulk flow transfer – Fast, long-distance – Chem nature of drug not impt • Short distances – Diffusion – Chem properties impt

Chem Properties Impt to Diffusion • Aqueous diffusion delivers most drug mol’s • Rate of diffusion dependent on molec size – Diffusion coeff = 1/ q. MW – BUT: Most drugs 200 -1000 MW, so little difference • Ability to cross barriers – Cell membr’s mostly lipid – Drug hydrophobicity impt

Absorption = Movement Across Cell Barriers • Cell membr’s separate aqueous compartments – Movement through cell involves traversing at least 2 lipid bilayers • Some tight junctions between cells – Ex: CNS, placenta, testes • Some freely permeable – Ex: Liver, spleen • Vascular endothelium differs in permeability

Small Mol’s Cross Cell Membr’s • Diff’n lipid • Diff’n aqueous pores traversing lipid bilayer – BUT most pores too small to accomm most drugs • Transmembr carrier prot • Pinocytosis – Not impt for small mol’s

Diffusion • Number mol’s crossing membr per unit area in unit time; depends on • Permeability coefficient (P) – Diffusivity • Diffusion coefficient • Doesn’t differ much between drugs – Solubility in membr • Partition coefficient (solubility oil/solubility water) • Most impt to pharmacokinetics • Used as predictor of drug properties

barbital secobarbital thiopental

p. H and Ionization • Many drugs are weak acids/bases – Can be ionized, unionized – Varies w/ p. H of environment • Acids release H+ – Strong: All H+ released – Weak: Some H+ released • Ka quantitates strength of acid
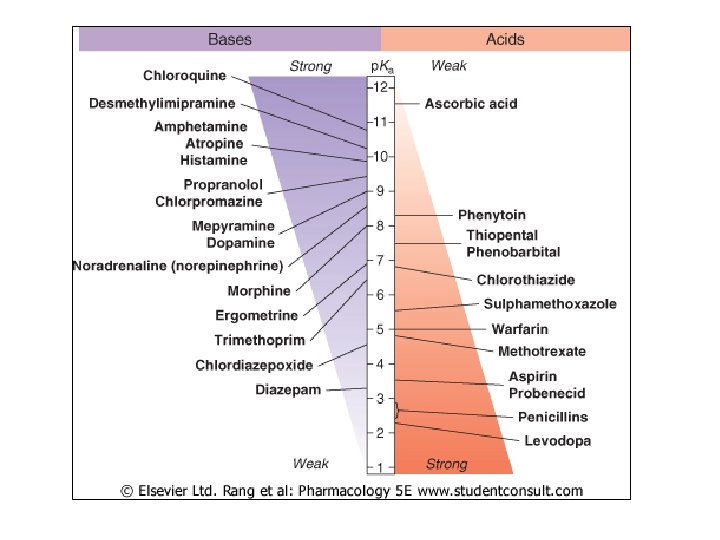
![• Weak acid ionization (HA H+ + A-) – Ka = [H+][A-]/[HA] –](http://slidetodoc.com/presentation_image/0cfc6e69ed0b68678988836eace7141a/image-12.jpg "• Weak acid ionization (HA H+ + A-) – Ka = [H+][A-]/[HA] –")
• Weak acid ionization (HA H+ + A-) – Ka = [H+][A-]/[HA] – Negative log and rearrangement: • Log [H+] = log Ka + log [A-]/[HA] – p. H = p. Ka + log [A-]/[HA] • Henderson/Hasselbach equation • p. Ka = p. H when drug 50% dissoc’d • Weak base ionization (BH+ H+ + B) – Ka = [H+][B]/[BH+] – Negative log and rearrangement – p. H = p. Ka + log [B]/[BH+]

• Rearrangement if known p. H, p. Ka allows deter’n ionized/unionized ratio at any p. H environment

p. H Differences between Body Compartments • Environmental p. H effects ability to release H+ (ionization) • Ionized species have low lipid solubility – Most: uncharged can traverse cell membr’s • So each environment’s p. H effects drug dist’n between them – Ion trapping – Compartment equilibrium
 w/ p. Ka=6. 0 •")
Ex: Stomach Blood • Assume weak acid drug (HA) w/ p. Ka=6. 0 • Assume [HA]=1. 0 • Stomach p. H=1. 0 – 1. 0 -6. 0=log [A-]/[HA] – 1. 0 x 10 -5=[A-] • Little ionized drug • Blood p. H=7. 0 – 10=[A-] • Much ionized drug • Expect stomach-to-plasma traverse BUT not plasma-to-stomach
 Plasma Digestive Tract")
Book ex: more basic drug (how do we know? ) Plasma Digestive Tract
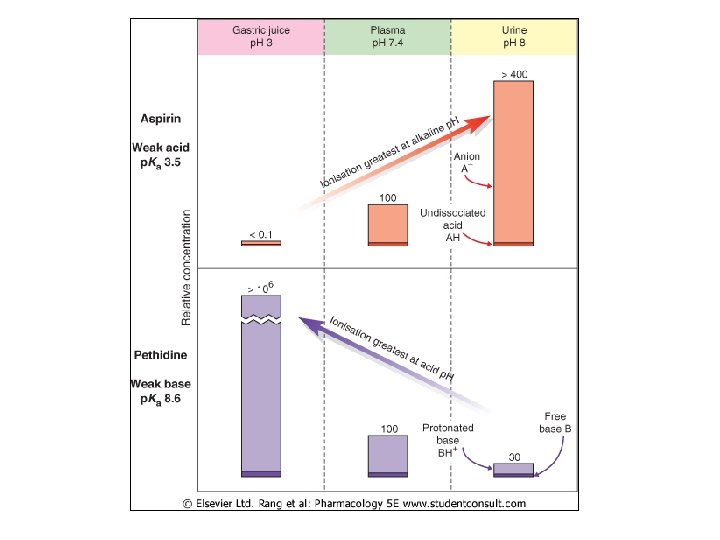
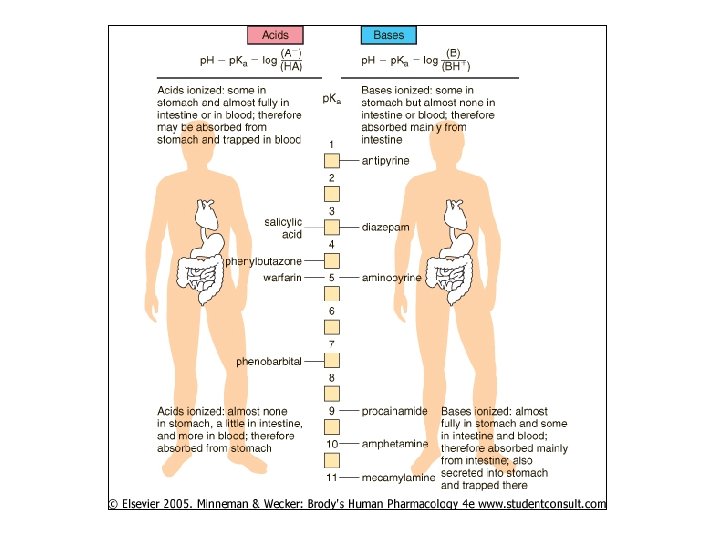

• Acidic drugs concent’d in high p. H compartment – Site of highst dissoc’n H+ (ionization) – Can’t traverse membr to escape • Basic drugs concent’d in low p. H environment • Largest D p. H between compartments largest D [drug] – BUT not total impermeability – AND not total equilib – Most impt to gi, renal

Carrier Mediated Transport • Specialized for physiologically impt mol’s – Sugars, neurotransmitters, metals, etc • Transmembr prot – Binds mol(s) – Changes conform’n – Releases to other side of membr • Diff kinetics than simple diffusion – Can become saturated – Subject to competition between ligands

• Two types of carriers allow – Facilitated diff’n • Along concent gradient – Active transport • Against gradient • Cell uses chem energy • Carriers impt pharmacologically – Renal tubule – Biliary tract – Blood-brain barrier – GI tract • P-glycoprotein impt drug transporter – Renal tubular cells, bile canaliculi, brain microvessels

Drug Administration
 • Oral • Sublingual")
Two Major Routes • Enteral – Via gastrointestinal tract (gi) • Oral • Sublingual • Buccal • Parenteral – Via injection • • IV IM Subcu Intrathecal

Oral Administration • Convenient; includes most drugs • Little absorption until small intestine – Are most drugs weak acids or bases? • Abs’n from small intestine – Passive transfer dependent on • Ionization • Lipid solubility – Some carrier-mediated transport • Levodopa through carrier for phenylalanine • Fluorouracil through carrier for pyrimidines • Fe, Ca

• Rates abs’n after oral admin depend on – Gi motility • Some disorders gastric stasis • Some drugs affect motility (incr or decr) • Meals – Splanchnic blood flow – Drug particle size/formulation • Capsules/coated tablets • Timed release formulations – Physicochemical factors • Tetracycline binds Ca milk prevents abs’n • Drug interactions

Bioavailability • Proportion of drug that passes into systemic circ’n after oral admin • Dependent on – Absorption – Local metab by small intestine enzymes • Indiv pts’ physiology impt – Activity intestinal metab enz’s – p. H variations – Motility
 – Oral dosing further metab •")
• Differs w/ type dose (oral, IV) – Oral dosing further metab • Book: First pass effect through liver – = AUCoral/AUCIV x dose. IV/doseoral • AUC = Area Under Curve of drug plasma concent vs. time • “Bioequivalence” used to compare generic drugs to patented

Other Types of Drug Admin • Sublingual – Impt when • Rapid response req’d • Drug unstable at gastric p. H • Drug rapidly metab’d by liver – Pass straight into systemic circ’n • Don’t enter liver portal system (so no first-pass effect)

• Ex: glyceryl trinitrate relieves angina – Metab NO release – NO act’s soluble guanylate cyclase (sim to ad cyclase) – incr’d c. GMP act’n prot kinase G – biochem cascade in smooth muscle – dephosph’n myosin light chains, sequestering Ca – vascular smooth muscle relaxation • Also relaxes cardiac muscle – decr’d bp, so red’d preload, cardiac afterload • So decr’d cardiac O 2 consumption – Also redist’n coronary blood flow toward ischemic cardiac areas

• Rectal – Abs’n unreliable – Often for local action – Useful in pts vomiting, unable to take by mouth (infants) • Cutaneous – Local effect on skin req’d – Abs’n occurs systemic effects – Suitable for lipid-soluble mol’s – Ex: estrogen patch

• Nasal sprays – Abs’n through mucosa overlaying lymphoid tissue – Impt for drugs inact’d in gi – Ex: peptide hormone analogs, ADH, calcitonin • Inhalation – Large surface area and high blood flow – No gi inact’n – BUT also route of elim’n – Ex: volatile, gaseous anesthetics – Ex: locally acting drugs – Ex: inhaled human insulin being tested

Admin by Injection • Subcutaneous, intramuscular – Faster than oral – Rate abs’n depends on site admin, local blood flow – Red’n or prolonging systemic action poss by altering drug mol or prep’n or giving w/ another agent • Intrathecal – Into subarachnoid space via lumbar puncture – Ex: regional anesthetics – Ex: cancer chemotherapeutics – Ex: antibiotics for NS infections
 fastest, most certain – Bolus high concent R heart, lung,")
• Intravenous (IV) fastest, most certain – Bolus high concent R heart, lung, systemic circ’n – Peak concent depends on rate injection – Common ex: antibiotics, anesthetics – Most uncomplicated to understand distribution, pharmacokinetics

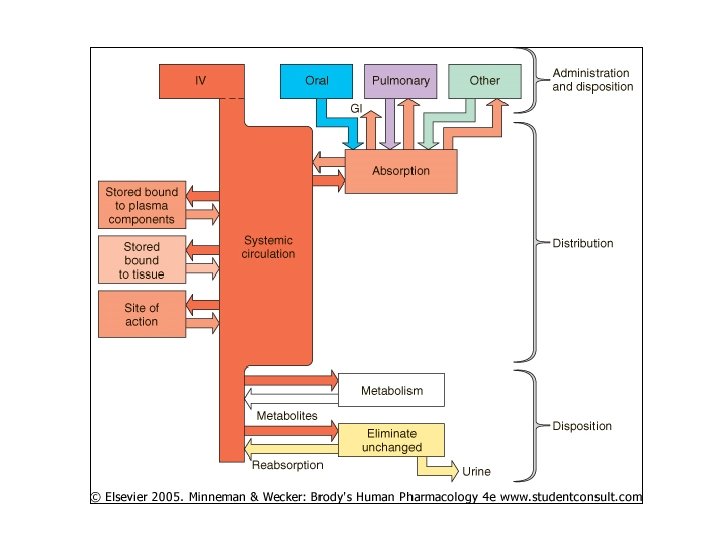
Distribution of Drugs in the Body Pharmacokinetics

Experimental Finding • Rates drug abs’n, dist’n, elim’n gen’ly directly proportional to physio concent • First order kinetics – Rate varies w/ first power of concent d. C(t)/dt = -k. EC(t) where d. C(t)/dt = rate change [drug] k. E = elimination constant (neg sign due to decr [drug] w/ elim’n) • Note: rate elim’n may be zero order (independent of concentration) – Ex: ethanol

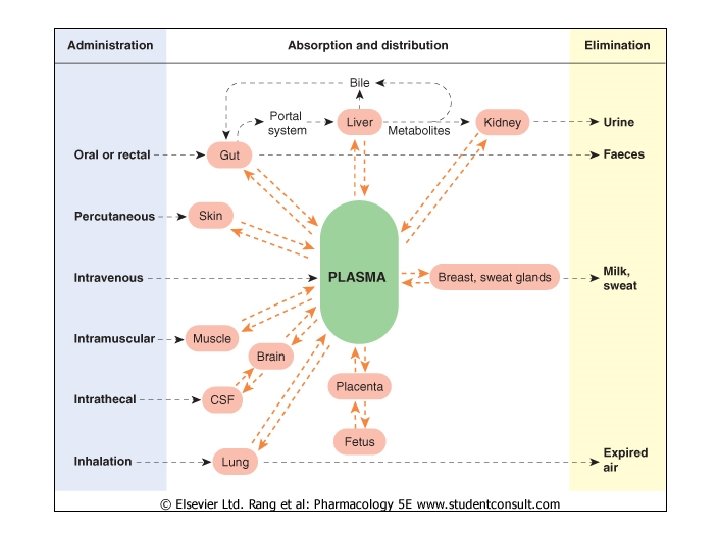
Kinetics Meas’d w/ Single IV Dose • Single bolus over 5 -30 sec • Periodic blood samples analyzed for [drug] – Time ~0 – highest concent • Dist’n drug in circulation equilib • Complete by sev passes through heart (sev min) – Later time – concent decr’s due to • • Dist’n tissues Dist’n other body fluids Metab other cmpds Excr’n unchanged drug (renal, biliary, lung)
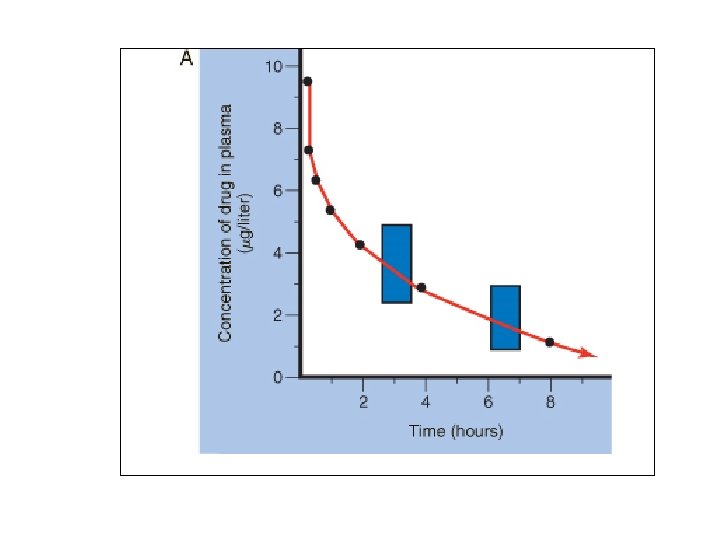
 reflects free drug + drug bound to plasma prot’s)")
• (Concent (y axis) reflects free drug + drug bound to plasma prot’s) • Conversion to log concent more linear curve – Non-linear portion – dist’n phase (a phase) • Rapid decr plasma concent – Linear portion – elimination phase (b) • Grad decr plasma concent
 = C 0 e-k. Et –")
• Eq’n line for elim’n phase: C(t) = C 0 e-k. Et – Where C(t) = Concent drug @ time (t) C 0 = Concent @ time 0 e = nat’l log base k. E = rate const for phase (elim’n rate const) t = time – Y int = C 0; slope = -k. E/2. 3 • Can be used to deter rate dist’n when a phase included
![With Oral Admin… • Plot differs in a phase • Initial: [plasma] = 0](http://slidetodoc.com/presentation_image/0cfc6e69ed0b68678988836eace7141a/image-40.jpg "With Oral Admin… • Plot differs in a phase • Initial: [plasma] = 0")
With Oral Admin… • Plot differs in a phase • Initial: [plasma] = 0 – Swallowing, dissolution, abs’n take time • Rapid abs’n rate b phase incr’s – First order: rate incr w/ incr’d [drug] • Peak concent at rate abs’n = rate elim’n
![Body Fluid Compartments: Sites of [Drug] • Total body water=50 -70% total body wt](http://slidetodoc.com/presentation_image/0cfc6e69ed0b68678988836eace7141a/image-41.jpg "Body Fluid Compartments: Sites of [Drug] • Total body water=50 -70% total body wt")
Body Fluid Compartments: Sites of [Drug] • Total body water=50 -70% total body wt • Intracell highest • Extracell: – Interstitial = between cells – Plasma = blood + lymph – Transcell = cerebrospinal, intraocular, synovial, etc. • Fat is also compartment – BUT poorly perfused
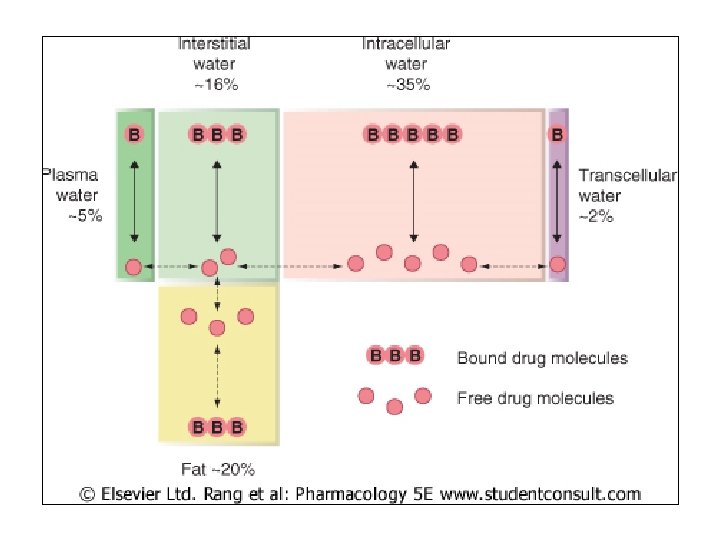

• Dug mol’s exist ionized/unionized, free/bound in each compartment • Dist’n pattern for each drug dependent on – Membrane permeability/transport – Binding w/in compartment – p. H partitioning – Fat/water partitioning

Specialized Compartment – Blood Brain Barrier • History: Ehrlich -- dyes injected IV stained most tissues; brain unstained • Contin layer endothelial cells w/ tight junctions – Non-brain – fenestrations • Specific transport for small organics • Safety buffer • Throughout brain, spinal cord – Except floor of hypothal, area postrema
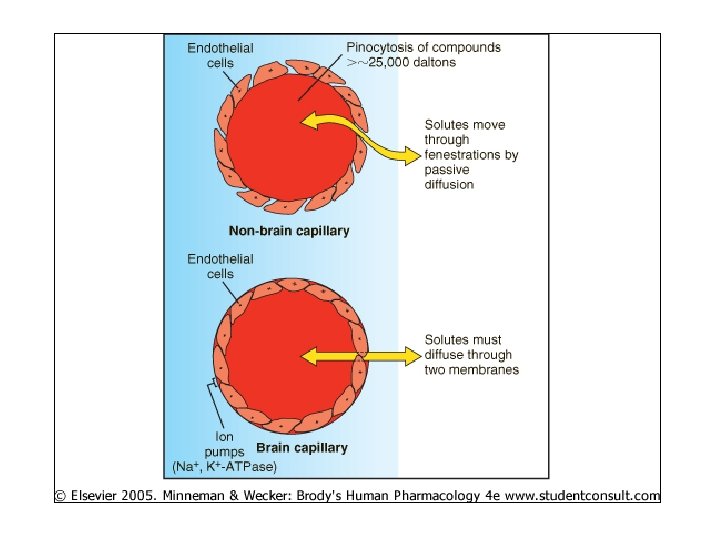

• Inaccessible to many drugs unless high lipid solubility – BUT inflamm’n can disrupt integrity – AND some peptides increase bbb permeability – Intrathecal injection sometimes circumvents

Volume of Distribution • Vol fluid req’d to contain drug in body at same concent as that present in plasma • May indicate drug binding to plasma prot or other tissue constituents • Vd = D/C 0 – Where Vd = vol dist’n (L) D = dose w/ IV injection (mg) C 0 = blood concent @ 0 time (mg/L)

• Most impt: free drug in interstitial fluid • Drug values vary greatly – Molecular wt – More impt: binding plasma prot’s

Drug Binding to Plasma Proteins • Reflected in Vd • If high binding, drug “trapped” in plasma – High C 0 on graph (Y reflects bound + unbound drug) – For Vd=D/C 0, Vd very low (2 -10 L) • Ex: warfarin (anticoagulant) • If low binding, drug free to disperse tissues – Low plasma concent (= low C 0) – Vd high (40, 000 L) • Ex: furosemide (diuretic)
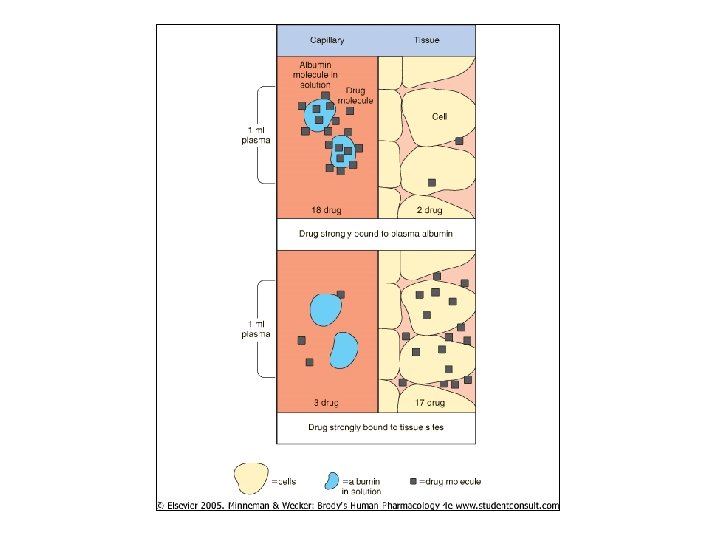

• Plasma proteins that bind drugs – Albumin impt to acidic drugs • Most abundant plasma prot – Not fully saturated • Synth’d in liver • Concent changes w/ disease, dysfunction – a-acid glycoprotein impt to basic drugs • Lower concent than albumin • Varies among population • Varies in individual if disease states – Lipoprotein binding not well understood • Varies w/ disease states

Clearance • Vol blood cleared of drug per time • Describes efficiency of elim’n from body – Sum of all types elim’n • Renal • Hepatic • Organ • Impt; independent of – Vol dist’n – Bioavailability – Half-life
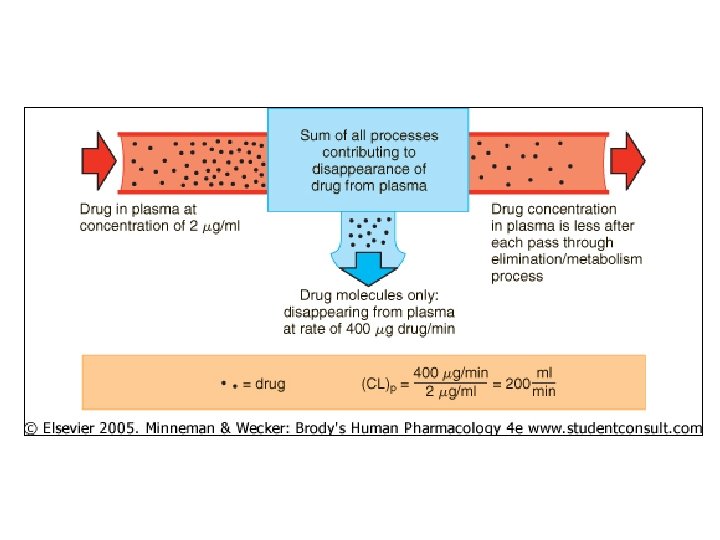

• Elim’n rate – quantity of drug removed – Assume first order kinetics • CLp = rate elim’n drug/plasma [drug] Where CLp = total body removal from plasma (p) (m. L/min), when rate elim’n (mg/min) plasma concent (mg/m. L) • Useful clin’ly for dosage rate, if target concent known
![Half-Life (t 1/2) • Time nec for [drug] to decr by half • Can](http://slidetodoc.com/presentation_image/0cfc6e69ed0b68678988836eace7141a/image-55.jpg "Half-Life (t 1/2) • Time nec for [drug] to decr by half • Can")
Half-Life (t 1/2) • Time nec for [drug] to decr by half • Can be found from graph log C(t) vs. t where C(t) = concent drug @ time t • Mins – days • Impt to deter’n multiple dosing regimen • Dependent on clearance, vol dist’n t 1/2 = (0. 693 x Vd)/CL

Drug Metabolism

Biotransform’n Drug Molecules • Drug changed chem’ly metabolite – Prodrugs must be metab’d for act’n • Chem alteration by enz rxn • Gen’ly nonpolar, lipid-sol cmpds more polar, water-sol – Now easier urinary excr’n • Some metabolites active (or more active) than parent drugs – Ex: demethlyation diazepam (less) active agent but w/ longer ½ life than parent

• Drug metabolizing enz’s mostly in liver • BUT most other tissues also can metabolize – Lung – Kidney – Gi – Placenta – Gi bacteria

• Four impt types chem rxns for drug metab – Oxidation – Reduction – Conjugation – Hydrolysis • Ox’n, conjugation most impt • Partic enz’s carry out these rxns

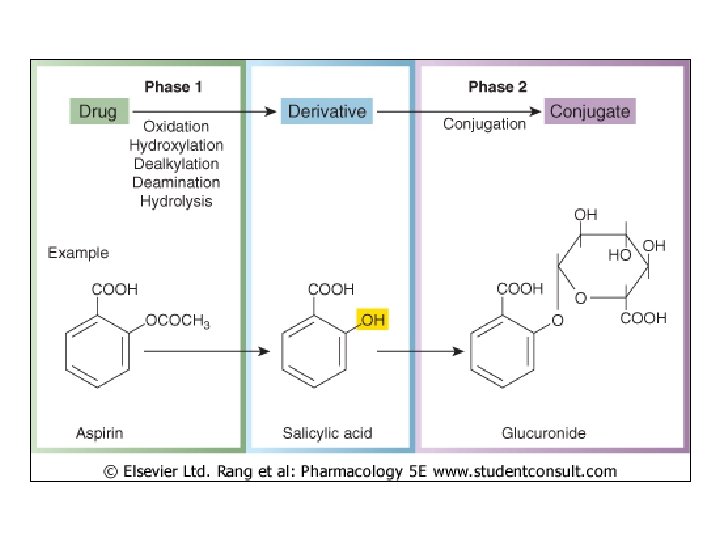
Two Major Metabolism Types • Phase I Reactions – Catabolic • Mostly ox’ns – Functionalization: • Intro reactive grp (ex: hydroxyl) • Prod’s more chem’ly reactive, hydrophilic than parent • Serves as pt chem attack for….
 – Involve conjugations rxns • Attachment")
• Phase II Reactions – Anabolic (synthetic) – Involve conjugations rxns • Attachment substituent • Large, hydrophilic • Liver major site Phase I, II rxns – Metabolic enz’s embedded in smooth ER • Microsomal • Stereoselective • Both types rxns more polar, hydrophilic metabolites
 – Enz superfamily •")
Phase I Rxns • Catalyzed by Cytochromes P 450 (CYP’s) – Enz superfamily • 74 CYP gene families • Differ in aa seq, inhibitors/inducers, specificity – 3 main families impt to hepatic drug metab (CYP’s 1, 2, 3) • CYP 1 A 2 – a main enz – Contains heme w/ Fe • Redox capability • Binds O 2 – Assoc’d w/ NAD(P) reductase enz • Allows metab many diff agents • Most common to all substrates: lipophilicity
H + H+ + O 2 DOH +")
• Gen’l rxn: DH + NAD(P)H + H+ + O 2 DOH + NAD(P)+ + H 2 O where DH = drug NAD(P)H = red’d coenzyme DOH = ox’d drug NAD(P)+ = ox’d coenzyme O 2 = final electron acceptor • Complicated cycle results in 1 O atom added to drug, other O water – Free radical or iron-radical grps formed at parts of cycle • Highly reactive, dangerous
")
Fp = Flavin Protein Coenzyme (NADPH-P 450 Reductase)
 include red’n, hydrolysis – Alcohol dehydrogenase")
• Other metabolic rxns (some enz catalyzed) include red’n, hydrolysis – Alcohol dehydrogenase metab’s ethanol – Monoamine oxidase metab’s many amines

• Some foods, other drugs, herbs, environmental agents, inhibit/induce CYP’s change in metabolism drugs change drug activity – Grapefruit juice, St. John’s wort inhibit drug metab by inhib’n CYP enz’s – Brusssels sprouts, cigarettes induce P 450 enz’s

Phase II Reactions • Attachment substituent grp on parent/ metabolite – Typically added @ hydroxyl, thiol, amino – Substituent first “activated” • Phosph’n • Att’d to Co. A • S-Adenosyl methionine • Rxn enzyme-catalyzed • Product almost always inactive, less lipid soluble – Excr’d in urine, bile

• Common conjugated substituents – – – Glucuronyl Sulfate Methyl Acetyl Glycyl Glutathione
![Rates of Drug Metab • Follow MM kinetics – V = (Vmax[S])/KM + [S]](http://slidetodoc.com/presentation_image/0cfc6e69ed0b68678988836eace7141a/image-72.jpg "Rates of Drug Metab • Follow MM kinetics – V = (Vmax[S])/KM + [S]")
Rates of Drug Metab • Follow MM kinetics – V = (Vmax[S])/KM + [S] • In vivo, Vmax directly proportional to [enz] – Can have competition between drugs metab’d by same enz – BUT most drugs found at concent’s << KM so far below saturation of enz active sites

• Enzyme induction – synth of metabolic enz’s stimulated – Both microsomal and conjugating systems – See incr’d metabolic activity • Due to repeated exposure to – Drugs – Environmental chem’s – Carcinogens • May decr’d drug activity OR incr’d activity

Figure 3 -10 Example of enzyme induction. Zoxazolamine administered by intraperitoneal injection to rats. For induction studies, phenobarbital or 3, 4 -benzo[a]pyrene was injected twice daily for 4 days before injection of zoxazolamine.
")
• Most thoroughly studied inducers – PAH’s (env chem’s also found in cigarettes) – Lipophilic – Bind nuclear receptors (Ah receptors) – Complexes bind response elements on DNA – Promotion transcr’n CYP 1 A 1 gene • Book: Induction P 450 s by PAHs in cigarette smoke decr’d estradiol in female smokers

First Pass Effect of Liver • Liver site of much metab – Amt abs’d >>> amt reaching systemic circ’n – Impt to many drugs • Much give much larger oral dose than needed • Indiv variations in extent of first-pass effect for partic drugs among population (so unpredictability)
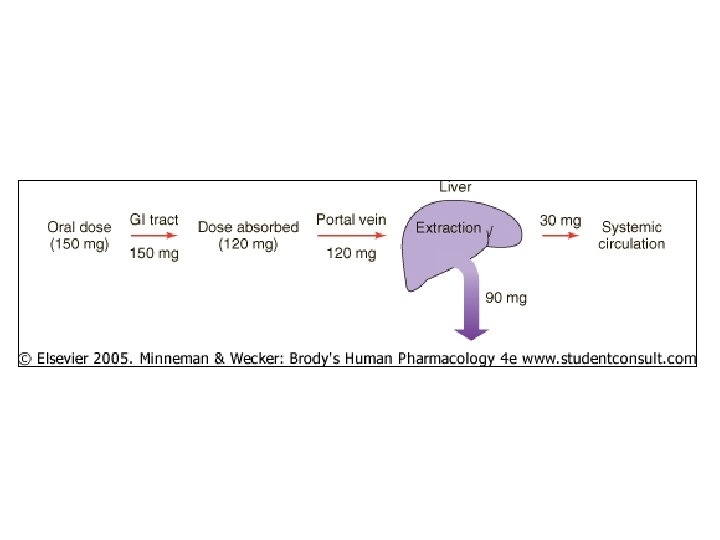
 impt")
Biliary Excr’n • Liver may excrete drugs bile – Drug carrier system (P-glycoprotein) impt • Bile duct carries small intestine • Glucuronides concent’d in bile – At small intestine, enz’s cleave glucuronide active drug released, reabs’d – Cycle repeated – “Enterohepatic circulation” – “reservoir” of recirculating drug

Elimination from the Body
, others")
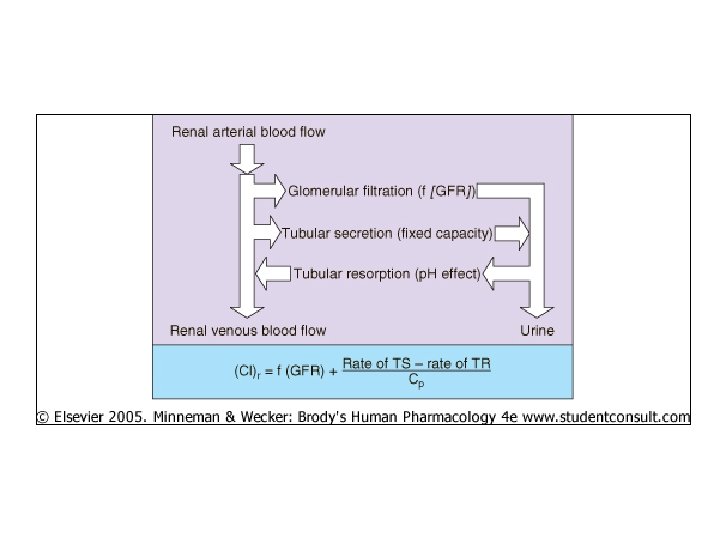
Renal Elimination • Kidney clears some drugs very efficiently (penicillin in single pass), others take many passes through renal tubule for excr’n • Glomerular filtration – ~20% renal plasma flow filtered through glomerulus – MW < 20000 diffuse into glomerular filtrate – Appreciable binding to albumin decr’d diffusion into filtrate

• Tubular secretion – ~80% renal blood flow passes through peritubular capillaries of prox tubule – Two carriers transport drugs from blood in capillaries proximal renal tubule • Carrier for acidic mol’s (including endogenous acids) • Carrier for basic mol’s • Move against electrochem gradient – Can achieve max drug clearance – Movement of free drug mol’s out of plasma pushes equilib toward freeing drug mol’s from albumin more free drug elim’d and more drug dissoc’ng from albumin

• Diffusion across renal tubule – Most water reabs’d from renal tubule • Concentrates urine – Highly lipophilic drugs can move across tubule cell membr’s, reabs’d back into blood – Highly polar drugs (low permeability) remain in tubule – p. H change in urine ionization trapping in urine elimination • Basic drugs more rapidly excr’d in acidic urine

One-Compartment Model • Simplest kinetics • Volume of distribution considered single, wellstirred compartment
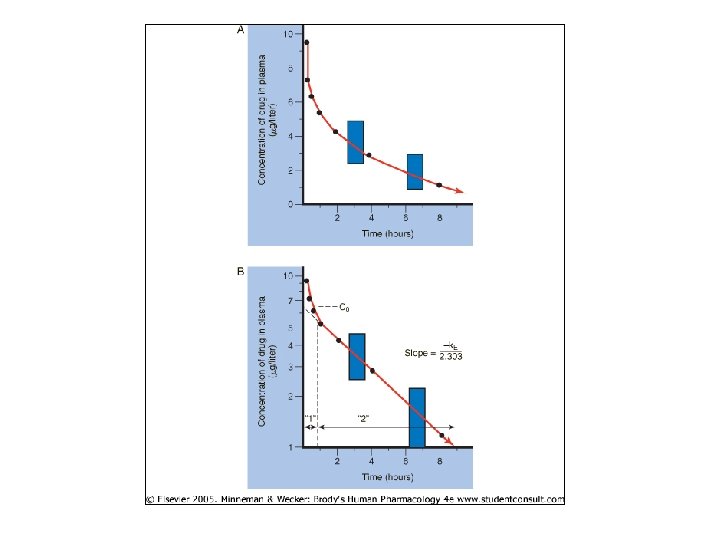

Real Life is More Complicated… Two Compartment Model
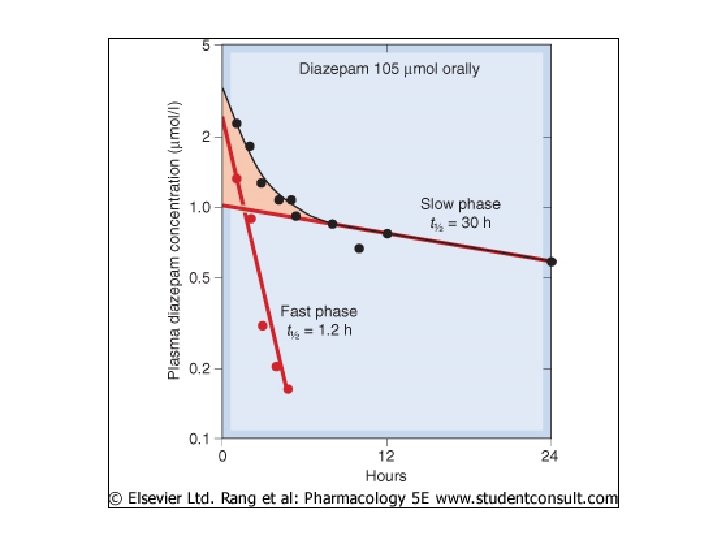

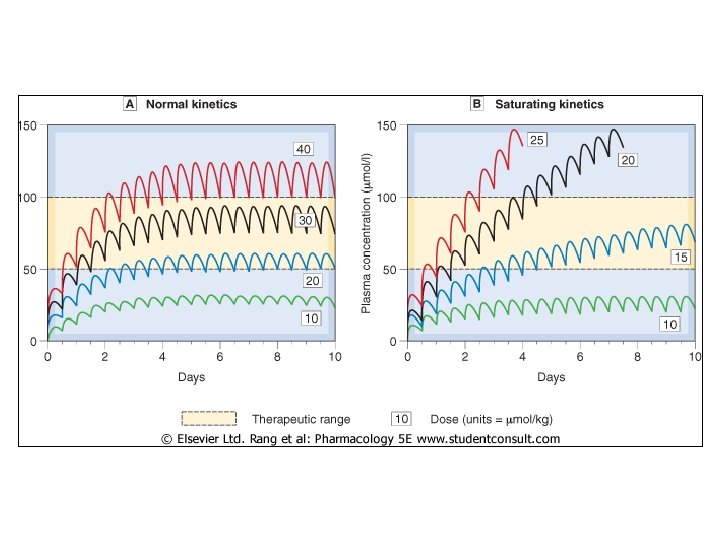
• Add repeated doses, saturating kinetics, other physiological parameters: kinetics more difficult
- Slides: 90