Down Syndrome Infantile Rhabdoid Tumour Hypothesis Objectives Rhabdoid

Down Syndrome Infantile Rhabdoid Tumour Hypothesis?

Objectives • Rhabdoid Tumour what is it? • What does “growing out of it” mean? • Examples of proliferative clinical phenomena during infancy and early childhood • Discuss hypotheses for childhood malignancies • Where does Down Syndrome cancer risk in childhood /adulthood fit in?

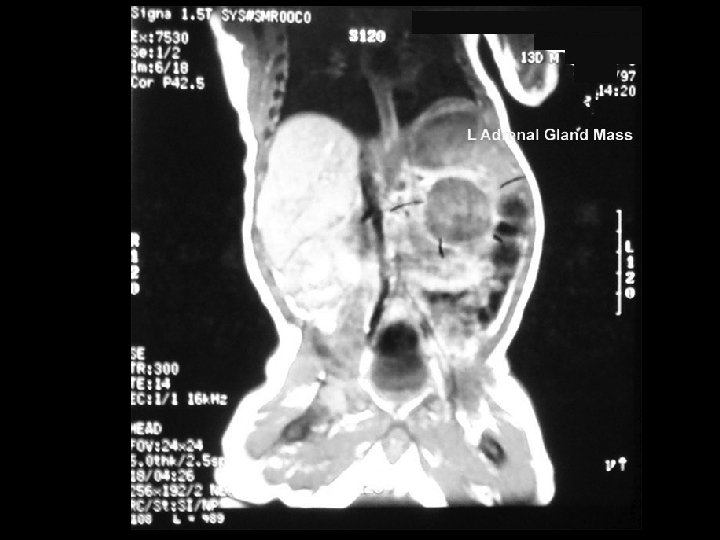
Infantile Malignant Tumour Incompletely resected

Infantile Malignant Tumour Incompletely resected

SMARC 1 b mutation present INI 1 stain negative Confirmatory of Rhabdoid Tumour

Rhabdoid Tumour Highly malignant tumour of infancy, involving the kidney, brain and other soft tissue sites (extra renal rhabdoid tumour) Rhabdoid cell mimic muscle cell precursor - embryonic Familial predisposiiton reported Associated with smarc 1 b germline mutation in a proportion of cases. Smarc 1 b mutation also identified in tumour tissue, yet not germ line Relentlessly progressive may be multi-focal – kidney and brain Treatment: Complete surgical resection, intensive chemotherapy and radiotherapy - frequently insufficient to control progression

Buzz What causes cancer? What do you know about physiology of Down Syndrome / trisomy 21 that may affect cancer development / risk?

All Cancer Age Incidence SEER Database

Growth Velocity Chart

ALL CNS Bone STS GCT - CNS

CNS Tumours Age Incidence SEER Database

Conclusion Ontogeny of hepatic GH-IGF axis: Prenatal: – IGF- I, -II and PRL receptors and IGF-I show a gestational increase Postnatal: – Loss of IGF-I, IGF-II and PRL receptors up to 6 months after birth – No significant change in GH receptor or IGF-I or -II m. RNA Loss of IGF and PRL receptors may be more important in determining the onset of GH dependent growth than alterations in GH receptor per se.

Transient Abnormal Myelopoiesis • Indistinguishable from AML M 7 • Exclusively associated with trisomy 21 • +++ Increased subsequent risk of AML (M 7), ALL, MDS
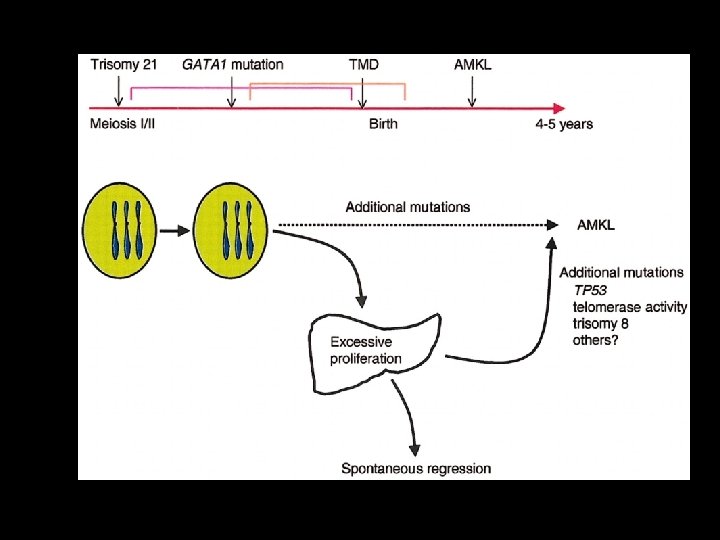
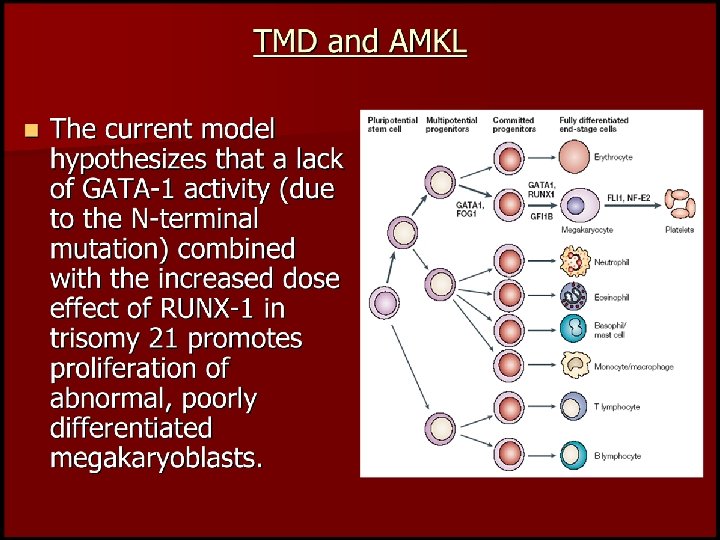
 A Developmental Hypothesis Scotting Perilongo & Walker Nature Reviews")
Solid Tumours of Childhood (CT) A Developmental Hypothesis Scotting Perilongo & Walker Nature Reviews Cancer (2005) 5; 481 -488 • Age /development-specific vulnerability suggested by age incidence patterns of CTs reflect peaks in growth in host tissues and vulnerability to oncogenic hits. • CTs are therefore unavoidable complications of growth and development. • Studying these mechanisms may provide important clues to novel therapies.

Solid Tumours of Childhood A Developmental Hypothesis Scotting Perilongo & Walker Nature Reviews Cancer (2005) 5; 481 -488 • Embryonic / Fetal Tissue Fidelity individual cell biology : microenvironment. • Teratocarcinomas arise in multiple tissues; demonstrate multiple phenotypes; yet revert to non-malignant phenotype when transplanted back to embryo (Martin 1981, Andrews 2002) • Tumour growth arrest or involution clinical phenomena Infantile Haemangioma Infantile Cardiac Rhabdomyoma of Tuberous Sclerosis Infantile arrest of myeloproliferative syndrome in Down’s Infantile Neuroblastoma 4 s Infantile fibrosarcoma, Arrested progression of infantile LCH Arrested tumour development / progression retinoblastoma Arrested tumour growth pilocytic astrocytoma

Cancer and Down Syndrome Leukaemias TAM / AML M 7 ALL: less frequent in infancy, T cell, TELAML 1, hyperdiploid, t(4: 11) and t(9: 22) less frequent - ? Due to Tiam 1, upregulated in trisomy 21 hypothesised.
, Wilms Tumour")
Cancer and Down Syndrome Childhood Solid Tumours Neuroblastoma (s 100 b differentiation), Wilms Tumour and CNS Tumours negative association Germ Cell Tumours and Retinoblastoma positive association

Cancer and Down Syndrome Adult Cancers 50% decreased risk – breast cancer almost absent, also reduced risk for genito-urinary tumours, lung cancer, oesophageal ca. ? Low environmental exposures ? Shortened life span Candidate tumour suppressor genes linked to trisomy 21 including Copper Zinc Superoxide Dismutase, Collage XVII, endostatin, calcineurin inhibition – antiangiogenesis

Cancer and Down Syndrome Worthy of further investigation What is your favoured hypothesis?
- Slides: 25