Distribution of Drugs Dr Naila Abrar Learning objectives

Distribution of Drugs Dr. Naila Abrar

Learning objectives After this session, you should be able to: > recall distribution of total body water > define distribution > define volume of distribution (vd) > explain the significance of vd > explain the factors affecting drug distribution > describe plasma protein binding and its effect on vd

Distribution? Where do drugs go?

Relative size of various distribution volumes within a 70 -kg individual

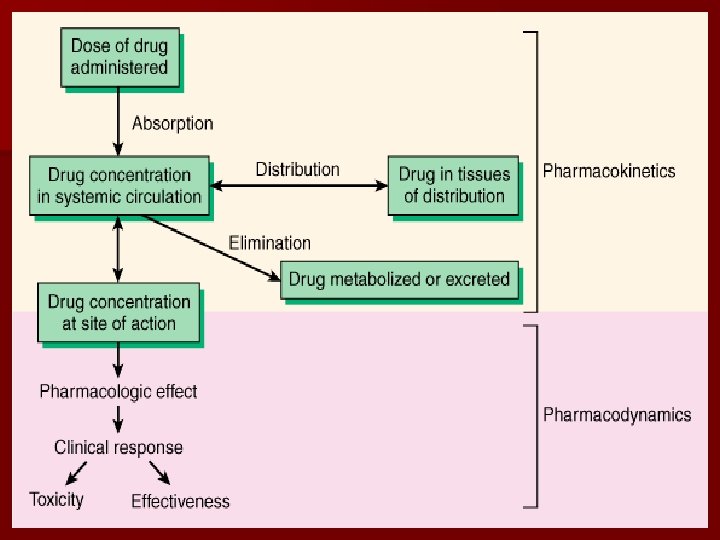
Definition Drug distribution is the process by which a drug reversibly leaves the bloodstream and enters the extracellular fluid and/or the cells of the tissues
 relates the amount of drug in the")
Volume of Distribution Volume of distribution (Vd) relates the amount of drug in the body to the concentration of drug (C) in blood or plasma Vd = Amount of drug in body Concentration

n An apparent and hypothetical volume in which the drug is distributed in the body n Reflects the extent to which the drug is present in the extra vascular tissue and not in the plasma

Significance n Why is it relevant ? To calculate the loading dose of a drug Ø loading dose= Vd x desired concentration Dialysis Ø Large Vd means the drug is in the peripheral compartment and thus not easy to be dialyzed

n Vd plasma concentration loading dose n Rapid absorption relative to the distribution plasma concentration can be higher than desired and may result in severe toxicity. Ø e. g. lidocaine n So, rate of administration can be crucial

One Compartment model For a first order elimination process, the graph of log Cpt versus time should give a straight line with a slope characterized by the elimination rate constant. Ø This is the case if the drug is distributed only in the plasma compartment. Ø

Two Compartment model Ø In practice, for most drugs, the decay curve is characterized by two or more elimination rate constants and the graph shows two phases: à phase of rapid decline à phase of slower decline

Plasma Concentration Profile after a Single I. V. Injection

Factors affecting distribution of drugs A. Blood flow B. Tissue permeability of the drug C. Binding of drugs to tissue proteins D. Binding of drugs to plasma proteins

Tissue permeability of the drug § Physiochemical Property: • Molecular size • Lipid solubility • Degree of ionization • Partition coefficient • Tissue specific transporters § Physiological barriers: • Blood Brain Barrier • Passage across Placenta

Binding of drug to tissue component n Binding of drug to plasma protein – Plasma protein binding • albumin • alpha-acid glycoprotein – Binding with blood cells • Hb: phenytoin, phenobarbitol • Carbonic anhydrase: acetazolamide • Cell membrane of RBC: imipramine

n Binding of drug component with tissue & extra vascular component: Ø Ø Ø Ø Liver : Paracetamol, chloroquine, digoxin Skin : Chloroquine Eye : Ephedrine, atropine Bones & teeth : Tetracycline, phenytoin Fat : DDT, thiopental, minocycline Skeletal muscle, heart : digoxin, emetine Brain : acetazolamide, chlorpromazine Kidney, vestibular apparatus : gentamicin

n Drugs binding to specific intracellular organelle – Mitochondria: tetracycline – Nuclei: chloroquine n Fat as a reservoir – Lipid soluble drugs

Plasma Protein Binding Albumin Globulin Free drug Bound drug Class I drugs Class II drugs
 Ø Liver")
n Increased Vd Ø Pregnancy Ø Renal failure (due to fluid retention) Ø Liver failure (due to altered body fluid & plasma protein binding) n Decreased Vd Ø Dehydration Ø Older mass) age (decrease in skeletal muscle

Redistribution n Initially distributed to organs with high blood flow n Later to less vascular but more bulky tissues n Greater the lipid solubility of the drug, faster is its redistribution n Termination of drug action, if the site of action was in one of the highly perfused organs (thiopentone sod. )

Thank you !
- Slides: 22