Cytogenetika lovka Autozomov aneuploidie Trizomie 21 Downv syndrom
 L. Down (1866) - nejčastější chromozomová")
 Edwards 1960 četnost 1: 8000 průměrná doba přežívání 2 měsíce")
 Patau 1960 86% postižených dětí umírá v prvním roce života.")
 Turner (1938) - Ford (1959) – 45,")
 popsán H. Klinefelterem (1942) Jacobsová a Strong (1959) – konstituce")

 popsáno Lejeunem")


 – „dynamická mutace“ např. u Huntingtonovy chorey, myotonické dystrofie cytologicky se")


 DNA hypersenzitivní ke klastogenům")

- Slides: 17
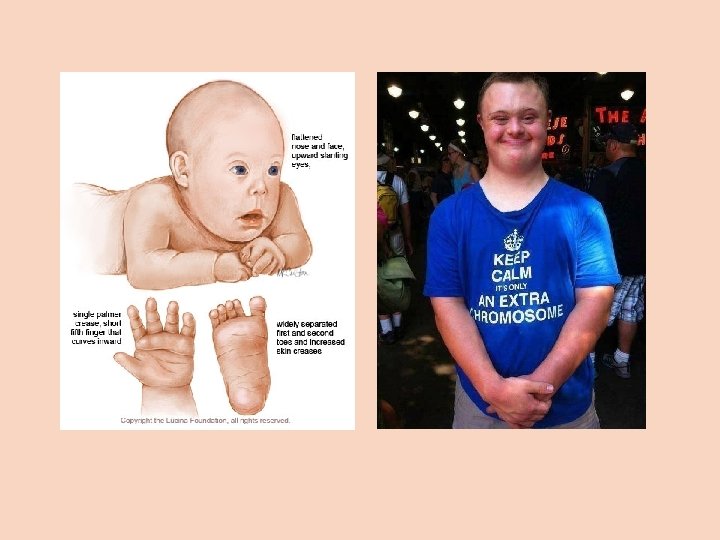
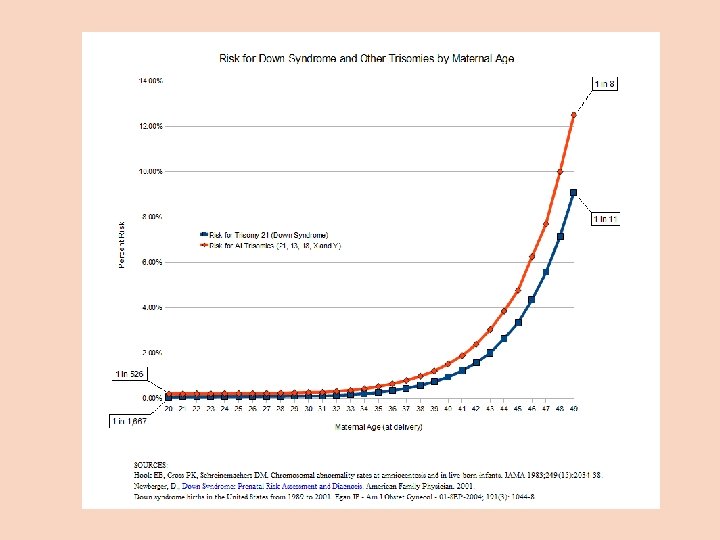
Cytogenetika člověka Autozomové aneuploidie Trizomie 21 (Downův syndrom) L. Down (1866) - nejčastější chromozomová odchylka u člověka, výskyt 1: 800 neobvyklá distribuce v populaci: vyšší věk matky a konkordance u monozygotních dvojčat: taková distribuce by mohl být odrazem chromozomové anomálie (Waardenburg, 1932) potvrzeno v r. 1959 riziko leukémie zvýšeno 15 x
Trizomie 18 (Edwardsův syndrom) Edwards 1960 četnost 1: 8000 průměrná doba přežívání 2 měsíce 80% ženského pohlaví vyšší věk matek
Trizomie 13 (Patauův syndrom) Patau 1960 86% postižených dětí umírá v prvním roce života. Frekvence 1: 4000 (10 000)
Aneuploidie pohlavních chromozomů Monozomie X (Turnerův syndrom) Turner (1938) - Ford (1959) – 45, X frekvence jen 1: 10 000 naopak nejčastější chromozomová odchylka u spontánních potratů (18% chromoz. abnormalit u spontánních potratů ) mozaicismus mnohem častější než u odchylek počtu autozomů (u 40% případů) jen 60% klasický 45, X: v ostatních případech strukturní změny chromozomu X či mozaicismus Inteligence normální nebo jen mírně narušená, Verbální IQ v normálu, nižší je ale performační IQ Poruchy prostorového vnímání estrogeny – druhotné pohl. znaky, růstový hormon
Syndrom XXY (Klinefelterův syndrom) popsán H. Klinefelterem (1942) Jacobsová a Strong (1959) – konstituce pohlavních chromozomů XXY vyšší věk matky – většinou nondisjunkce chromozomů X mateřského původu (60%) ostatní případy nondisjunkce chromozomů X a Y v prvním meiotickém dělení na rozdíl od Turnerova syndromu není diagnostikovatelný před pubertou podnětem pro vyšetření je většinou sterilita pacienti vysocí a štíhlí, s dlouhými končetinami menší obvod hlavy již při porodu performační IQ je téměř normální, nižší je ale verbální IQ, problémy s učením spíše pasivní a submisivní ve srovnání s vrstevníky vnímavost k sociálnímu stresu
Syndrom XYY frekvence 1: 1000 nondisjunkce Y v druhém meiotickém dělení otce vysocí muži, IQ kolísá: v některých případech IQ sníženo jindy normální, plodnost normální nebo jen mírně snížená poruchy s učením, hyperaktivita, agresivita později často poruchy sociální přizpůsobivosti: agresivní nebo psychopatické chování Syndrom XXX frekvence 1: 1000 ženská „analogie“ Klinefelterova syndromu vyšší, štíhlé ženy s dlouhými končetinami, už v dětství podstatně menší obvod hlavy opožděný rozvoj řeči častá oligofrenie (IQ 55 -75), zejména nižší je verbální IQ, potíže s učením a v mezilidských vztazích ve 2/5 je narušena funkce pohlavních žláz nebo se objevuje přímo neplodnost, v ostatních případech je plodnost normální nebo jen mírně snížená plodné ženy netransmitují syndrom XXX Syndromy XXXX a XXXXX Těžká retardace somatického a psychického vývoje, u pentazomie navíc mnohočetné vývojové anomálie
Chromozomové aberace Delece delece na krátkém raménku chromzomu 5 (cri du chat) popsáno Lejeunem a kol. frekvence výskytu 1: 50 000 – 1: 100 000 velká a plochá obličejová část, malá mozkovna, typický pláč epikantus, antimongoloidní průběh očních štěrbin, nízko posazené uši které mají někdy výrůstky, ustupující brada, snížené IQ Delece na krátkém raménku chromozomu 4 (Wolfův syndrom) Mikrocefalie s těžkými poruchami vývoje CNS Často rozštěp patra, deformace skeletu, kraniální asymetrie Abnormální genitál, vrozené vady srdce dysmorfie ledvin – podkovovitý tvar
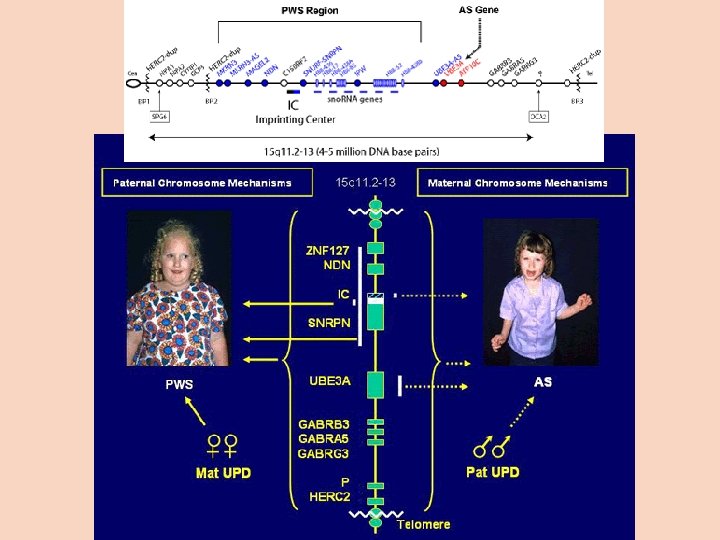
Mikrodelece Syndrom Prader-Willi krátká delece na dlouhém raménku chromozomu 15 zděděná od otce útlé dětství: porucha pití, zvláštní pláč nebo pláč chybí kryptorchismus, opožděný vývoj a pozdní nástup řeči, okrouhlá, hypomimická tvář dětství: hyperfagie, obezita, nízká postava, krátké ruce a nohy adolescence: poruchy chování, sekundární pohlavní znaky se objevují opožděně nebo se vyvinou jen neúplně stejná delece zděděná od matky: Angelmanův syndrom retardace psychomotorického rozvoje, těžká porucha řeči, záchvaty smíchu, křečí hypertonie končetin x hypotonie trupového svalstva Retinoblastom Delece na dlouhém raménku chromozomu 13 hereditární forma – heterozygoti Rb – nádory většinou na obou očích izolovaná forma – většinou jen jeden nádor Di Georgeův syndrom – dlouhá raménka chr. 22, aplazie thymu CATCH 22
Inverze Pericentrická inverze na chromozomu 3 newfoundlandský rodokmen děti s rekombinovaným chromozomem (duplicita části dlouhého ramene a chybění části krátkého ramene): ustupující brada, mentální retardace, anomálie srdce a ledvin Pericentrická inverze na chromozomu 8 jihozápad USA Traslokace Některé Robertsonovy přestavby jsou častými populačními polymorfismy v lidské populaci, zejména mezi chromozomy 13 a 14 a mezi 14 a 21. Translokace mezi chromozomy 13 a 14 je zřejmě nejčastější chromozomovou mutací našeho druhu (1: 1300). Přes absenci fenotypových projevů problémy: produkce nebalancovaných gamet. U translokace zahrnující chromozom 21 zvýšené riziko potomka s Downovým syndromem.
Expanze trinukleotidů (1991) – „dynamická mutace“ např. u Huntingtonovy chorey, myotonické dystrofie cytologicky se manifestuje jen v někt. případech, např. na chromozomu X – syndrom fragilního X (FRAXA) neboli Martinův-Bellův syndrom, 1: 1250 (muži), 1: 2000 (ženy) druhá nejčastější chromozomová příčina mentální retardace mužů (po Downově syndromu) vyšší postava, dlouhá tvář s vystouplou bradou, velké odstávající uši, velké testes (zejména v pubertě), mentální retardace (IQ 35 -70), opožděný nástup řeči a její poruchy, častý autismus přenašečky mohou být fenotypově zcela normální, třetina je ale lehce mentálně retardovaná nebo má alespoň problémy s učením fragilní místo leží v subtelomerické oblasti dlouhého ramene chromozomu X
cytologická detekce problematická: u postižených mužů je fragilní X viditelný jen v cca 35% buněk, u přenašeček ještě v nižším procentu buněk procento buněk s markerem je možno zvýšit zavedením speciálních kultivačních podmínek, tedy média s nízkou koncentrací kyseliny listové a thymidinu, popř. po aplikaci fluordeoxyuridinu nebo methotrexátu proto se dnes FRAXA detekuje převážně molekulárně biologickými metodami (Southern s použitím metylsenzitivních restiktáz nebo PCR). kromě FRAXA na chromozomu X i další fragilní místa, zejména běžné FRAXD a vzácné FRAXE a FRAXF. Další místa byla zjištěna na autozomech. Je zajímavé že další fragilní místa (kromě FRAXE) nejsou spojena s patologickým fenotypem. FRAXA nemá typický obraz dědičnosti markeru vázaného na chromozom X (tzv. paradox Shermanové) 20% přenašečů (mužů) fenotypově normálních. Dcery těchto mužů taky asymptomatické. 40% vnuků ale postižených. Z vnuček může mít až čtvrtina mírnou mentální retardaci. Molekulární podstata onemocnění: expanze trinukleotidů CGG v promotoru genu FMR 1 (norm. počet opakování 5 -58, patologický 230 – 2000). Při velkém rozsahu expanze jsou trinukleotidy metylovány (ve variabilním rozsahu) K expanzi a následné metylaci dochází jen u přenašeček
Syndromy zvýšené lámavosti chromozomů poruchy reparace DNA Xeroderma pigmentosum řídké onemocnění, v evropské populaci frekvence 1: 250 000 autozomově recesivní onemocnění heterogenní choroba: 80 -90% porucha excizní reparace poškození DNA vyvolaného uv zářením. 10 -20% defekt postreplikační reparace DNA. Čtyři stadia onemocnění: 1. Difúzní pigmentace po expozici slunečnímu záření 2. Intenzivní pigmentace, suchá pokožka, teleangiektazie 3. Prekancerozní stadium 4. Neoplastické stadium, neurologické poruchy 2/3 pacientů umírají do 20 let na malignity a sekundární infekce mírný průběh onemocnění – pacienti se dožívají středního věku
Fanconiho anémie autozomově recesivní dědičnost, vzácné onemocnění (1: 350 000) DNA hypersenzitivní ke klastogenům fenotyp velmi variabilní: obligátně pancytopenie v řadě případů (50%) poruchy růstu, mikrocefalie, vývojové anomálie konců končetin, u 20% mentální poruchy, hluchota. Dále časté poruchy pigmentace (hypo- nebo hyper). Pacienti většinou umírají v mládí (krvácení, infekce, leukémie) Ataxia teleangiectasia Autozomově recesivní dědičnost (1: 40 000 až 100 000) ATM gen – lokalizován na dlouhém ram. chromoz. 11 - odpověď buňky na genotoxický stres v interakci s proteinem p 53. Hypersenzivita na ionizační záření, teleangiektázie (nejdříve na očních spojivkách), ataxie, imunodeficity, častý výskyt rakoviny
Bloomův syndrom autozomově recesivní dědičnost, 1: 100 000 časté u Židů skupiny Aškenázi mutace genu pro DNA ligázu I (na dlouhém rameni chromozomu 15) teleangiektatický erytém motýlovitého tvaru s typickou lokalizací v obličeji porucha růstu, imunodeficity, zvýšený výskyt malignit Diagnostika: neobyčejně zvýšená frekvence SCE, DNA diagnostika Cockayneův syndrom autozomově recesivní dědičnost hypersenzitivita na uv záření nanismus, stařecký vzhled obličeje (v důsledku ztráty podkožního tuku), progresivní mentální retardace, intrakraniální kalcifikace, porucha sluchu, retinitis pigmentosa a atrofie očního nervu, fotosenzitivní dermatitida disproporcionálně dlouhé končetiny s velkými rukami a nohami