CURSO DE RESIDNCIA MDICA E PSGRADUAO ONCOLOGIA HEMATOLOGIA


 Coagulação Trombose")






 é um fenômeno de")

 Ø ü ü ü Dçs. Linfoproliferativas:")
 revestem a superfície das")






— equimoses nas pernas Petequias Escala atual:")













 Tipo 1 • É o tipo mais comum, entre 60 -80% dos")
 Tipo 2 • É um defeito qualitativo e acomete entre 20 -30%")

 Tipo 3 • É o tipo mais grave caracterizado pela deficiência total")








 T. T. P. A. (normal) Erro de")
 T. P. Normal Prolongado Erro Laboratorial Mistura")









- Slides: 61
CURSO DE RESIDÊNCIA MÉDICA E PÓS-GRADUAÇÃO ONCOLOGIA - HEMATOLOGIA HIAE Síndromes Hemorrágicas Dr. João Carlos de Campos Guerra Departamento de Patologia Clínica - HIAE Centro de Hematologia de São Paulo - CHSP
SISTEMAS ENVOLVIDOS NA HEMOSTASIA • Hemodinâmico • Vascular • Plaquetário • Coagulação • Fibrinolítico • Inibidores
SISTEMAS ENVOLVIDOS NA HEMOSTASIA Hemodinâmico Hemorragia Fibrinólise Fator Vascular Plaquetário Equilíbrio (Hemostasia) Coagulação Trombose Inibidores Anticoagulantes
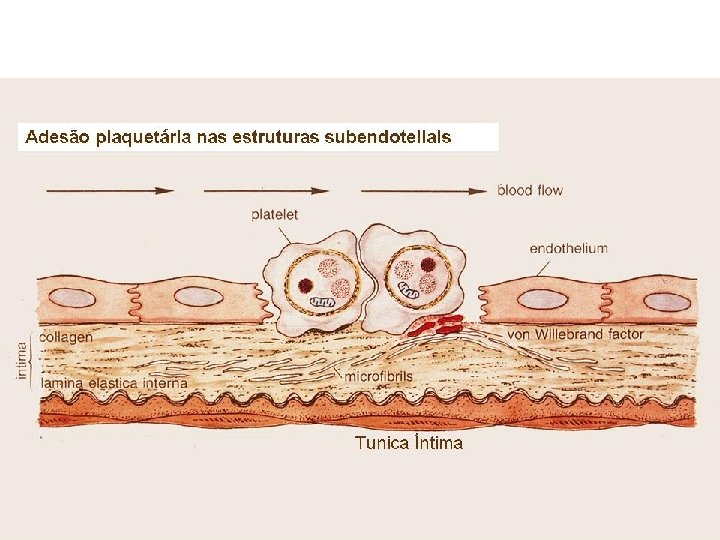
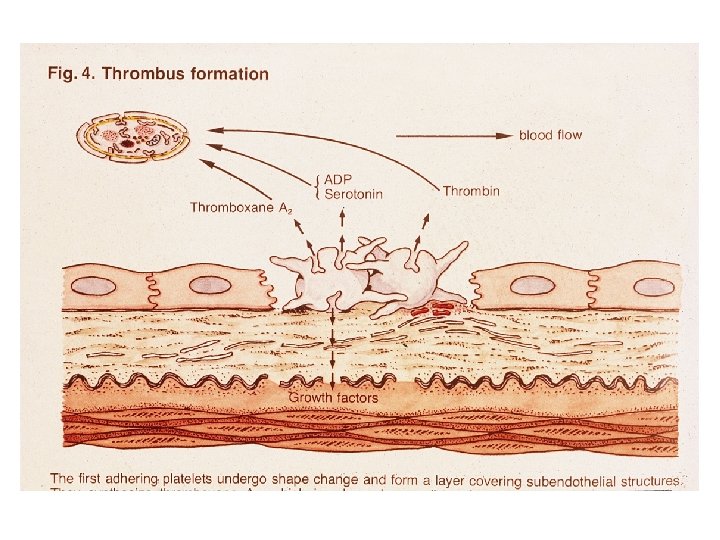
HEMOSTASIA PRIMÁRIA Heparinase Fator Va PDGF Tromboxane A 2 Fibronectina Formação do Trombo: Fibrinogenio Fv. W adesão ativação agregação 2. ATIVAÇÃO/SECREÇÃO 1. ADESÃO 3. AGREGAÇÃO
V Va Fase de Iniciação X VIII/ Fv. W II VIIIa Vlla Va Xa TF IIa Célula que expressa FT Vlla TF IX VIIIa plaqueta II X Xa IIa XI Va IXa Plaqueta ativada Fase de propagação Xla
End'othelium TF TISSUE FACTOR PATHWAY INHIBITOR F XIIa PK HMWK F Vlla F IX F Xla Ca++ PL Extrinsic X-ase Complex F l. Xa Intrinsic X-ase Complex Ca++ PL F Vlll Figure 1 Clotting cascade: calcium ions; phospholipids (PL); and a substrate SYMBOL. TF, tissue factor; PK, prekallikrein; HMWK, highmolecular-weight kininogens. From: Amitrano: Semin Liver Dis, Volume 22(1). February 2002. 83 -96 F Vl. Ila F Xa FV FX Prothrombin F Va Prothrombinase Complex Ca++ PL Thrombin F Xlll Fibrinogen F Xllla Crosslinked Fibrin polymer
F Vllla F Va Prothrombin Protein S Protein C Thrombomodulin Fibrin monomer Heparan Sulfate FPA FPB Fibrinogen Figure 2 Clotting pathway inhibitors and fibrinolytic system. SYMBOL, inhibitors; activators. FPA, fibrinopeptide A; FPB, fibrinopeptide B; t. PA, tissue plasminogen activator; PAI-1, plasminogen activator inhibitor 1; PAI-2, plasminogen activator inhibitor 2; TAFI, thrombin activatable fibrinolysis inhibitor. From: Amitrano: Semin Liver Dis, Volume 22(1). February 2002. 83 -96 Antithrombin III Thrombin Fibrin polymer Crosslinked Fibrin Fragment X. Y. D. E Fragment X. Y. E D-Dimer Plasmin Urokinase Alpha-2 -antiplasmin PAI-1 t-PA PAI-2 Plasminogen TAFI
TROMBOCITOPENIA Outras Causas Produção Deficiência da MO Eritropoiese Ineficaz Destruição Transfusão Maciça Hiperesplenismo Não Imunológica Aplasia QT Anemia Megaloblástica CIVD PTT RT SHU Drogas Alo Infecção Vasculites (TIH) Imune Toxina CEC Drogas Infiltração Trombocitopenias Hereditárias Auto Imune PTI LES P Neonatal LP PPT HIV Fig. Causas da trombocitopenia: MO= medula óssea, QT= quimioterapia, RT= radioterapia, CIVD= coagulação intravascular dissemidada, PTT= púrpura trombocitopênica trombótica, SHU= síndrome hemolítico-urêmica, P Neonatal= púrpura neonatal, PTT= púrpura pós transfusional, PTI= púrpura trombocitopênica imunológica, LES= lúpus eritematoso sistêmico, LP= doenças linfoproliferativas, HIV= vírus da imunodefifiência adquirida, HCV= hepatite por vírus C, TIH= trombocitopenia induzida por heparina. HCV
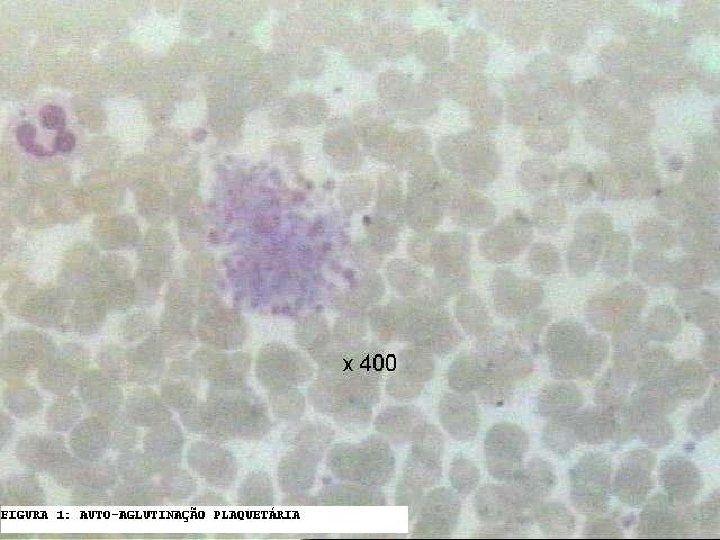
Diagnóstico reais e artefatos em Plaquetopenia • Em 45. 000 amostras analisadas, a prevalência de PTCP foi de 46 casos (0, 1%). A avaliação microscópica do “esfregaço a fresco” (sem anticoagulante) é útil para identificar a PTCP. – Batels PC, et al: Screening for EDTA-dependnet deviations in platelet counts and abnormalities in platele distribution histogram in pseudothrombocytopenia. Scand J Clin Lab Invest 1997 Nov, 57(7): 629 -36.
Diagnóstico reais e artefatos em Plaquetopenia • A pseudotrombocitopenia (PTCP) é um fenômeno de falsa baixa contagem de plaquetas (falsa plaquetopenia), ocasionado pela aglutinação “in vitro” das plaquetas na presença de autoanticorpos plaquetários e anticoagulantes, sendo mais comum o EDTA. A falha no reconhecimento desta alteração pode resultar em diagnósticos errôneos e tratamentos inapropriados. - Lombarts AJ, et al: Accurate platelet counting in an insidious case of pseudothrombocytopenia. Clin Chem Lab Med 1999 Nov-Dec, 37(11 -12): 1063 -6. - Ahn HL, et al: EDTA-dependent pseudothrombocytopenia confirmed by supplementation of kanamycin; a case report. Korean J Intern Med 2002 Mar, 17(1): 65 -8. - Schrezenmeier H, et al: Anticoagulant-induced pseudothrombocytopenia and pseudoleucocytosis. Thromb Haemost 1995 Mar; 73(3): 506 -13.
Trombocitopenia Imune Diagnóstico de exclusão Ø Hemograma normal exceto isolada plaquetopenia Ø Plaquetas gigantes no esfregaço, sem outras citopenias Ø Exame físico normal exceto sinais de sangramento Ø PTI secundária: ü Dç. Auto-imune ü Dç. Linfoproliferativa ü Infecção
Trombocitopenia Imune secundária Ø Auto-imune: ü Lupus (LES) Ø ü ü ü Dçs. Linfoproliferativas: LLC LÑH Macroglobulinemia de Waldenstron Ø ü ü Infecções: HIV HCV H. Pylori Dengue ü Sindrome do anticorco antifosfolípides (SAFI) ü Dç. Tireoidiana Imune (Graves) ü Sindrome de Evans
Patogênese da PTI Destruição das Plaquetas 1, 2 Autoanticorpos (imunoglobulinas) revestem a superfície das plaquetas Os receptores Fc dos macrófagos ligam-se a anticorpos na superfície das plaquetas As plaquetas são prematuramente destruídas sendo fagocitadas no baço Produção inadequada das Plaquetas 1 -4 Autoanticorpos se ligam a megacariócitos na medula óssea. Os megacariócitos são destruído ou apresentam maturação prejudicada O “turnover” das plaquetas pode estar diminuído, mesmo com níveis de TPO normais References: 1. Chang M, et al. Blood. 2003; 102: 887 -895. 2. Cines DB, et al. Annu Rev Med. 2005; 56: 425 -442. 3. von dem Borne, et al. Blood Rev. 2002; 16: 57 -59. 4. Mc. Millan R, et al. Blood. 2004; 103: 1364 -1369.
Fisiopatologia da PTI SRE Macrófago P Autoanticorpo plaquetário P Sangue Periférico P Medula Óssea P Megacariócito Plaqueta
MO – PTI X 10
MO – PTI X 10
MO – PTI X 50
MO – PTI X 100
Achados Clínicos e Evolução da PTI Crianças 1 -5 Adultos 1 -5 Geralmente aguda, transitória Crônica na maioria dos pacientes (crônica ~10 – 20%) Frequentemente após infecção viral ou imunização Não possue relação direta com outras patologias Remissão espontânea 80%– 90% em 2– 8 semanas Remissão espontânea <10% Refratário em 15 – 30% Refratário em 30 – 50% Risco muito baixo de mortalidade ou sangramento severo Mortalidade 8 -16% (refratário); 5% de hemorragia fatal (aguda) References: 1. Stasi R, et al. Mayo Clin Proc. 2004; 79: 504 -522. 2. Cines DB, et al. Annu Rev Med. 2005; 56: 425 -442. 3. George JN, et al. Blood. 1996; 88: 3 -40. 4. Kumar M, et al. Am J Hematol. 2005; 78: 181 -187. 5. Cines DB, et al. Blood. 2005; 106: 2244 -2251.
Sinais e sintomas de trombocitopenia Purpura (manchas roxas)— equimoses nas pernas Petequias Escala atual: 1 cm 2 cm 3 cm
Sinais e sintomas de trombocitopenia Sangramento anormal e hemorragia Sangramento anormal de mucosa RNM - hemorragia em SNC pac. com trombocitopenia severa
PTI – TRATAMENTO CRIANÇA Ø Resolução espontânea na maioria dos casos Ø Sangramento severo é incomum (KÜhne et al, Lancet 2001) Ø Frequência de PTI crônica 20 -25% Ø Trombocitopenia crônica severa é raro: < 5% (Blanchette, 2002)
PTI – TRATAMENTO ADULTO Ø Geralmente é uma condição crônica 2 Ø Incidência: ~50 to 100 por milhão ano in US, Europa 3 Ø Afeta todas as idades; a incidência dobra em mulheres adultas 1, 3 Ø Remissão espontânea: 5 - 11% Ø Sangramento severo é incomum cont. de plaq. > 30. 000/mm 3 (Cortelazzo et al, Blood 1991) Ø ü ü ü Risco de hemorragia fatal – cont. plaq. < 30. 000/mm 3 0, 4% ao ano em pacts < 40 anos 1, 2% ao ano em pacts 40 -60 anos 13% ao ano em pacts > 60 anos References: 1. Stasi R, et al. Mayo Clin Proc. 2004; 79: 504 -522. 2. Chang M, et al. Blood. 2003; 102: 887 -895. 3. Cines DB, et al. Annu Rev Med. 2005; 56: 425 -442. (Cohen et al, arch Intern Med 2000) 4. George JN, et al. Blood. 1996; 88: 3 -40.
NÚMERO DE PLAQUETAS TROMBOCITOPENIA E RISCO HEMORRÁGICO 1, 2 5% 20– 50, 000/mm 3 10% 10– 20, 000/mm 3 21% <10, 000/mm 3 0 5 10 15 20 25 RISCO DE HEMORRAGIA References: 1. Elting LS, et al. J Clin Oncol. 2001; 19: 1137 -1146. 2. Weaver CH. http: //patient. cancerconsultants. com/supportive_treatment. aspx? id=995. 2005.
PTI – TRATAMENTO QUAL PACIENTE DEVE SER TRATADO ?
PTI – TRATAMENTO ADULTO Ø Consenso: ü Observação em pacientes sem sangramento e cont. de plaq. > 30. 000/mm 3 Ø Drogas de 1 a linha: ü Corticosteróides ü Imunoglobulina ü Anti-D Ig Ø Tto. de 2 a linha: ü Esplenectomia Ø Pacientes refratários: ü Rituximab Ø Outras opções terapêutica: ü Vincristina, ciclosporina, azatioprina, danazol Ø Terapias experimentais: ü Receptores agonistas de trombopoetina
PTI – TRATAMENTO ADULTO DOSES Ø Doses de corticoesteróides: Ø Dose de Ig. IV: ü Alta: metilprednisolona (30 mg/Kg/dia) ü Baixa (0, 8 g/kg/dia) -3 dias ü “Standard”: (4 mg/kg/dia)- 7 dias ? ? ? ü Baixa: (2 mg/kg/dia) – 21 dias ? ? ? Ø Anti-D Ig IV: ü Dose de 50 -75 mcg/kg ü Duração da resposta 1 -5 semanas ü Alta (1 g/kg/dia) ü Regimes de 1 a 2 dias ü Duração da resposta 2 -4 semanas
Coagulopatias Hereditárias e Adquiridas HEMOFILIA Ø A: ↓ FVIII Ø B: ↓ FIX Ø C: ↓ FXI Severa: fator abaixo de 1% do normal. Moderada: fator entre 1 a 5% do normal. Leve: fator entre 5 a 40% do normal. Hemofilia C A hemofilia C, também conhecida como Síndrome de Rosenthal, é decorrente da falta do fator XI, envolvido na via intrínseca da cascata de coagulação. É um tipo de hemofilia que costuma ser leve, mas pode assemelhar-se à hemofilia clássica. Acomete ambos os sexos. É achado frequente entre famílias judias descendentes de linhagens de judeus Asquenazi. Difere da hemofilia A e B pois não leva a sangramentos nas juntas. Costuma cursar com sangramentos tardios em pós-operatório. É uma doença de herança autossômica dominante.
Coagulopatias Hereditárias e Adquiridas HEMOFILIA Alterações Laboratoriais ↑ TTPA e ↓ do fator Pesquisa de anticoagulante circulante negativa TTPA nl → deficiência TTPA↑→ inibidor
Coagulopatias Hereditárias e Adquiridas ALTERAÇÕES QUANTITATIVAS DOS FATORES DE COAGULAÇÃO Ø deficiência isolada de qualquer fator fibrinogênio, TP, TTPA e TT FVII : TP Ø deficiência múltipla → FV e FVIII
Coagulopatias Hereditárias e Adquiridas Doença de von Willebrand Ø autossômica Ø QC variável Ø disfunção plaquetária Ø ↑TS, ↑ TTPA, ↓ FVIII Ø ↓ agregação com ristocetina Ø ↓ Fv. W e do cofator da ristocetina
Doença de Von Willebrand • Doença Autossômica Dominante; • Alteração no fator VIII da coagulação, pois a subdivisão pró-coagulante é carreada pelo fator de von Willebrand (adesão plaquetária);
Sintomas • Sangramentos de leve a moderado que variam com a intensidade da doença; • Hematomas; • sangramentos menstruais prolongados; • sangramentos nasais; • sangramentos excessivos após pequenos cortes; • Gengivorragia, equimoses facéis.
Classificação a) Tipo 1 • É o tipo mais comum, entre 60 -80% dos casos; • É um defeito quantitativo onde a concetração do Fv. W fica entre 20 -50% do valor normal; • Causa sangramentos de leve a moderado.
Classificação b) Tipo 2 • É um defeito qualitativo e acomete entre 20 -30% dos casos. • 2 A: subtipo mais comum neste caso o Fv. W tem dificuldade de unir-se às plaquetas e há diminuição da presença de grandes multímeros.
Classificação • 2 B: o Fv. W tem grande afinidade de união às plaquetas, por isso diminui a circulação livre do Fv. W. • 2 M: não há ausência dos grandes multímeros porém eles não tem a mesma capacidade de ligação às plaquetas. • 2 N: o Fv. W perde a capacidade de ligação com o Fator VIII.
Classificação c) Tipo 3 • É o tipo mais grave caracterizado pela deficiência total do Fv. W. O paciente tem sangramentos severos.
Diagnóstico • Levar em conta a presença dos sintomas; • História familiar; • Estudo laboratorial.
FVIII Coag Fv. W: Ag Fv. W: Rco Agregação 1 ↓ ↓/N 2 A ↓/N ↓/N ↓ 2 B ↓/N ↓/N ↓ 2 M ↓/N ↓ ↓↓ ↓/N 2 N ↓↓ N N N 3 <5% <5%
Testes específicos • Dosagem de antígeno de von Willebrand é feita por ensaio imunológico (boa sensibilidade para os tipos 1 e 3 ); • Agregação induzida pela ristocetina; • Determinação dos múltimeros.
Coagulopatias Hereditárias e Adquiridas ALTERAÇÕES QUANTITATIVAS DOS FATORES DE COAGULAÇÃO ADQUIRIDAS insuficiência hepática Ø deficiência da vitamina K → F II, VII, IX e X Ø anticoagulantes Ø sangramento maciço Ø inibidores
Coagulopatias Hereditárias e Adquiridas ANTICOAGULANTES Ø heparina Ø antagonistas da vitamina K Ø venenos
Coagulopatias Hereditárias e Adquiridas HEMOFILIA ADQUIRIDA Ø hemofílicos Ø doenças auto-imunes Ø gestantes Øidosos
Coagulopatias Hereditárias e Adquiridas DEFEITOS QUALITATIVOS DOS FATORES Hereditários Ø disfibrinogenemia Adquiridos Ø CIVD
Coagulopatias Hereditárias e Adquiridas CONCLUSÕES Ø importante história pessoal e familiar Ø quadro clínico: tipo de sangramento Ø comorbidades Ø seqüência laboratorial de investigação com aprofundamento gradativo
EXAME ALTERADO T. T. P. A. (alterado) T. T. P. A. (normal) Erro de Laboratório Prolongado (mistura com PN) CORRIGE Deficiência NÃO CORRIGE Anticoagulante circulante > a incubação 2’, 5’, 10’ CORRIGE Fase Contato PROLONGADO VIII IX XI XII Anticoagulante lúpico (verificar trombose) antifosfolipideos não lupico hemorragia
EXAME ALTERADO T. P. PROLONGADO (Fora ) T. P. Normal Prolongado Erro Laboratorial Mistura CORRIGE Deficiência F. VII NÃO CORRIGE Anticoagulante Anti Vit. K inibidor
T. P. e T. T. P. A. ALTERADOS REPETIDOS NORMAL PROLONGADO Mistura CORRIGE I Baixo NÃO CORRIGE Anticoagulante Circulante Normal Lúpico X, V, II CID Fibrinólise Hipo Disfibrinogenemia Não Lúpico
Alterações da Coagulação 1. 2. 3. 4. TTPA Prolongado TP Normal TP Prolongado TTPA Normal TTPA Prolongado TP Prolongado TTPA Normal TP Normal deficiência de XII, XI, IX, VIII deficiência de VII deficiência de X, V, II, I paciente normal e deficiência XIII
Interpretação dos testes de triagem nos distúrbios hemostáticos TP TTPA TT PLAQ TS CONDIÇÕES N N N Consumo: PTI, CIVD Produção: leucemia, aplasia, drogas N N N Deficiência F XIII N N Alteração da função plaquetária: hereditária ou adquirida N N Deficiência vit. K, FVII, hepatopatia, ACO, inibidor N N Deficiência pré-calecreína, CAPM, FXII, FXI, FIX, FVIII, Dç. v. W, hepatopatia, anticorpo circulante, heparina N N N Deficiência congênita FX, FV, hepatopatia N N Def. /disfunção fibrinogênio, dç. hepática aguda, heparina
ESTUDO DA FUNÇÃO CAPILAR E PLAQUETÁRIA • Prova do Laço • TS de IVY • Agregação Plaquetária • Platelet Function Analyser - PFA 100
TS- Tempo de Sangramento
Agregação Plaquetária ADP – ADN – Ácido Aracdônico - Colágeno
PFA - 100 Fluxo com alto “shear stress” (pressão do deslocamento da lâmina de sangue circulante)
Tromboelastograma
F I M DR. JOÃO CARLOS DE CAMPOS GUERRA Departamento de Patologia Clínica-HIAE Centro de Hematologia de São Paulo jcguerra@einstein. br