CSCE 555 Bioinformatics Lecture 6 Sequence Alignment part
 Meeting: MW 4: 00 PM-5:")
CSCE 555 Bioinformatics Lecture 6 Sequence Alignment (part. III) Meeting: MW 4: 00 PM-5: 15 PM SWGN 2 A 21 Instructor: Dr. Jianjun Hu Course page: http: //www. scigen. org/csce 555 University of South Carolina Department of Computer Science and Engineering 2008 www. cse. sc. edu.

Roadmap Hashing Function based quick search Heuristic algorithm: FASTA, BLAST Multiple Sequence Alignment algorithm: Clustal W Summary 1/11/2022 2

Hash Table for Quick Search Smith 18 Alice 19 Bob 18 Lucy 28 Alicia 32 Dan 30 Ron 32 Georg e 32 O(n) O(log(n)) O(1)

Searching Consider the problem of searching an array for a given value ◦ If the array is not sorted, the search requires O(n) time If the value isn’t there, we need to search all n elements If the value is there, we search n/2 elements on average ◦ If the array is sorted, we can do a binary search A binary search requires O(log n) time About equally fast whether the element is found or not ◦ It doesn’t seem like we could do much better How about an O(1), that is, constant time search? We can do it if the array is organized in a particular way 4

Hashing Suppose we were to come up with a “magic function” that, given a value to search for, would tell us exactly where in the array to look ◦ If it’s in that location, it’s in the array ◦ If it’s not in that location, it’s not in the array This function is called a hash function because it “makes hash” of its inputs 5
 Hashing Function A hash function is a function that: ◦ When applied to")
(Magic) Hashing Function A hash function is a function that: ◦ When applied to an Object, returns a number ◦ When applied to equal Objects, returns the same number for each ◦ When applied to unequal Objects, is very unlikely to return the same number for each Hash functions turn out to be very important for searching, that is, looking things up fast 6
 hash function Suppose our hash function gave us the following values: hash.")
Example (ideal) hash function Suppose our hash function gave us the following values: hash. Code("apple") = 5 hash. Code("watermelon") = 3 hash. Code("grapes") = 8 hash. Code("cantaloupe") = 7 hash. Code("kiwi") = 0 hash. Code("strawberry") = 9 hash. Code("mango") = 6 hash. Code("banana") = 2 0 kiwi 1 2 3 banana watermelon 4 5 6 7 8 9 7 apple mango cantaloupe grapes strawberry
 { int")
Example of Hash Function PRIVATE int hash_number (const char *key, int size) { int hash = 0; ◦ if (key) { const char * ptr = key; ◦ for(; *ptr; ptr++) hash = (int) ((hash*3 + (*(unsigned char*)ptr)) % size); ◦ } ◦ return hash; }
 9")
FASTA (Fast Alignment) 9
 Approach (BLAST) (Altschul et al. 1990, developed by")
BLAST (Basic Local Alignment Search Tool) Approach (BLAST) (Altschul et al. 1990, developed by NCBI) ◦ View sequences as sequences of short words (k-tuple) DNA: 11 bases, protein: 3 amino acids ◦ Create hash table of neighborhood (closely-matching) words ◦ Use statistics to set threshold for “closeness” ◦ Start from exact matches to neighborhood words Motivation ◦ Good alignments should contain many close matches ◦ Statistics can determine which matches are significant Much more sensitive than % identity ◦ Hashing can find matches in O(1) time ◦ Extending matches in both directions finds alignment 10
 11")
BLAST (Basic Local Alignment Search Tool) 11

Multiple Sequence Alignment containing multiple DNA / protein sequences Look for conserved regions → similar function Example: #Rat #Mouse #Rabbit #Human #Oppossum #Chicken #Frog ATGGTGCACCTGACTGATGCTGAGAAGGCTGCTGT ATGGTGCATCTGTCCAGT---GAGGAGAAGTCTGC ATGGTGCACCTGACTCCT---GAGGAGAAGTCTGC ATGGTGCACTTGACTTTT---GAGGAGAAGAACTG ATGGTGCACTGGACTGCT---GAGGAGAAGCAGCT ---ATGGGTTTGACAGCACATGATCGT---CAGCT 12

Multiple Sequence Alignment: Why? Identify highly conserved residues ◦ Likely to be essential sites for structure/function ◦ More precision from multiple sequences ◦ Better structure/function prediction, pairwise alignments Building gene/protein families ◦ Use conserved regions to guide search Basis for phylogenetic analysis ◦ Infer evolutionary relationships between genes Develop primers & probes ◦ Use conserved region to develop Primers for PCR Probes for DNA micro-arrays 13

Multiple Alignment Model Q 1: How should we define s? X 1=x 11, …, x 1 m 1 Q 2: How should we define A? Model: scoring function s: A X 1=x 11, …, x 1 m 1 Possible alignments of all Xi’s: A ={a 1, …, ak} X 2=x 21, …, x 2 m 2 … Find the best alignment(s) XN=x. N 1, …, x. Nm. N Q 3: How can we find a* quickly? X 2=x 21, …, x 2 m 2 S(a*)= 21 … XN=x. N 1, …, x. Nm. N Q 4: Is the alignment biologically Meaningful? 14

Minimum Entropy Scoring Intuition: ◦ A perfectly aligned column has one single symbol (least uncertainty) ◦ A poorly aligned column has many distinct symbols (high uncertainty) 1/11/2022 Count of symbol a in column i 15
 columns are independent (2) linear gap cost =Maximum score")
Multidimensional Dynamic Programming Assumptions: (1) columns are independent (2) linear gap cost =Maximum score of an alignment up to the subsequences ending with Alignment: 0, 0, 0…, 0 ---|x 1| , …, |x. N| We can vary both the model and the alignment strategies NP-complete problem. High complexity 16

Approximate Algorithms for Multiple Alignment Two major methods (but it remains a worthy research topic) ◦ Reduce a multiple alignment to a series of pairwise alignments and then combine the result (e. g. , Feng. Doolittle alignment) ◦ Using HMMs (Hidden Markov Models) Feng-Doolittle alignment (4 steps) ◦ Compute all possible pairwise alignments ◦ Convert alignment scores to distances ◦ Construct a “guide tree” by clustering ◦ Progressive alignment based on the guide tree (bottom up) 17

Progressive Alignment

How to Align One Sequence to an Existing Alignment? Add a sequence to an existing group: a sequence s: CGAAATC want to align to a existing alignment s 1 s 2 AG–AT– -GAATC The high scoring pairwise alignment is s 2 s -G–AATC CGAAATC Hence , s is merged into the group alignment as: if needed add gaps s 1 s 2 s AG--AT– -G–AATC CGAAATC fixed

How to Align a Group to Another Group? Two groups: S 1 ATTGCCATT-- S 2 ATC-CAATTTT S 3 S 4 ATGGCCATT ATCTTC-TT The highest score alignment is S 1 – S 3 , so it is used for aligning the two groups as S 2 S 1 S 3 S 4 ATC–CAATTTT ATTGCCATT-ATGGCCATT-ATCTTC-TT--

Limitation of Feng-Doolittle Alignment Problems of Feng-Doolittle alignment ◦ All alignments are completely determined by pairwise alignment (restricted search space) ◦ No backtracking (subalignment is “frozen”) No way to correct an early mistake Non-optimality: Mismatches and gaps at highly conserved region should be penalized more, but we can’t tell where is a highly conserved region early in the process Iterative Refinement ◦ Re-assigning a sequence to a different cluster/profile ◦ Repeatedly do this for a fixed number of times or until the score converges ◦ Essentially to enlarge the search space 21

Clustal W: A Multiple Alignment Tool CLUSTAL and its variants are software packages often used to produce multiple alignments Essentially following Feng-Doolittle ◦ Do pairwise alignment (dynamic programming) ◦ Do score conversion/normalization (Kimura’s model) ◦ Construct a guide tree (neighbour-journing clustering) ◦ Progressively align all sequences using profile alignment Offer capabilities of using substitution matrices like BLOSUM or PAM Many Heuristics 22

One example of MSA using Clustalw

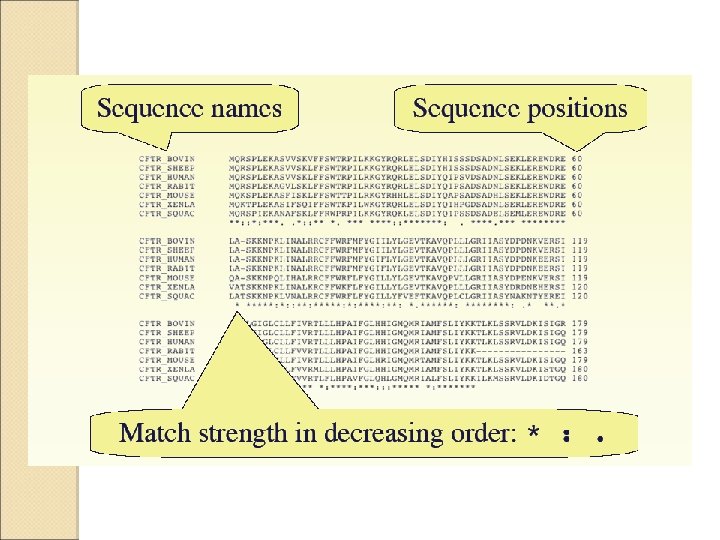
 MUSCLE stands")
More Advanced MSA algorithms Kalign MAFFT (Multiple Alignment using Fast Fourier Transform) MUSCLE stands for MUltiple Sequence Comparison by Log-Expectation. MUSCLE is claimed to achieve both better average accuracy and better speed than Clustal. W 2 or T-Coffee allows you to combine results obtained with several alignment methods

Measuring Alignment Significance The statistical significance of a an alignment score is used to try to determine if an alignment is the result of homology or just random chance. The E-value of an alignment score is the expected number of unrelated sequences in a database that would have a score at least as good. 27

E-values and p-values The E-value of a particular score is determined by multiplying the number of sequences in the database, n, times the p-value of the score. The p-value of score X is the probability of a single random alignment having score X or larger. E-value(X) = n • p-value(X) 28

Computing p-values To compute the p-value of X, we must know how random scores are distributed. The p-value of X is equal to the area under the distribution curve to the right of X. For ungapped local alignments, the distribution can be computed analytically. For gapped alignments, it must be estimated empirically. 29

Summary Hashing for quick search Blast and Fasta Progressive Multiple Sequence alignment Testing significance of alignments
 chapter 4 ◦ Textbook (EB)")
Next Lecture Profiles and HMM Reading: ◦ Textbook (CG) chapter 4 ◦ Textbook (EB) chapter 6
- Slides: 31