Cours dHmostase DUT ABB 1 Physiologie de lHmostase

Cours d'Hémostase DUT ABB 1 - Physiologie de l'Hémostase Auteur : Bruno Flamand, IUT de Dijon

PHYSIOLOGIE DE L’HEMOSTASE Ensemble des différents mécanismes assurant: -la prévention des saignements spontanés -l’arrêt des hémorragies en cas de lésion vasculaire -le maintien de la fluidité sanguine -une participation dans les phénomènes de cicatrisation

• L’Hémostase: 3 étapes inter-dépendantes qui se déroulent de façon concomitante - Hémostase primaire: aboutit formation d’un agrégat plaquettaire ( « thrombus blanc » ), permet seul l’arrêt des s aignements dans les capillaires les plus fins - Coagulation plasmatique: aboutit par la formation d’un réseau de fibrine, à la consolidation de l’agrégat plaquettaire - Fibrinolyse: permet la lyse du caillot fibrino-érythroplaquettaire, et le maintien de la perméabilité vasculaire, une fois la cicatrisation du vaisseau achevée

L’Hémostase: processus localisé et rapide, en équilibre physiologique entre processus coagulant et fibrinolyse, régulés eux-même par des inhibiteurs et des activateurs Plaquettes + Facteurs de la Coagulation Hémostase primaire Coagulation plasmatique Inhibiteurs physiologiques de la coagulation Activateurs du plasminogène Fibrinolyse Inhibiteurs physiologiques de la fibrinolyse Ce processus nécessite la coopération entre: -la paroi des vaisseaux sanguins -les protéines plasmatiques, facteurs de la coagulation et de la fibrinolyse -les cellules sanguines, en particulier les plaquettes
 UN TEMPS VASCULAIRE et")
HEMOSTASE PRIMAIRE Objectif : formation d’un agrégat plaquettaire (thrombus blanc) UN TEMPS VASCULAIRE et UN TEMPS PLAQUETTAIRE Facteurs mis en jeu: -paroi vasculaire -plaquettes -protéines plasmatiques

Déroulement de l’hémostase primaire LESION VASCULAIRE VASOCONSTRICTION ADHESION PLAQUETTAIRE ACTIVATION PLAQUETTAIRE AGREGATION PLAQUETTAIRE

1. VASOCONTRICTION : immédiate dès la rupture du vaisseau -Petits vaisseaux uniquement -Passive (élasticité paroi) puis active par contraction réflexe (sympathique) des muscles lisses -Prolongée et accrue par substances libérées par les plaquettes: adrénaline, sérotonine, TXA 2, … • Diminution jusqu’à 40% du Ø du vaisseau • Ralentissement du débit sanguin favorisant les interactions entre plaquettes et endothélium

2. ADHESION PLAQUETTAIRE au sous-endothélium vasculaire • Endothélium vasculaire « non thrombogène » : protéoglycanes tels que héparane sulfate, dermatane sulfate, … • Sous-endothelium « thrombogène » : collagène, microfibrilles, élastine, fibronectine • Plaquettes: glycoprotéines membranaires (GP) récepteurs - GP Ia/IIa : récepteur collagène - GP Ic/IIa : récepteur fibronectine - GP IIIb : récepteur thrombospondine - GP Ic’/IIa: récepteur laminine -GP Ib/IX : récepteur Facteur von Willebrand (v. WF) • Facteur von Willebrand: glycoprotéine (240 k. Da) plasmatique, synthèse par ¢ endothéliales et Mk, forme multimères (500 à 20 000 k. Da) 1 domaine de liaison aux fibres de collagène et 1 domaine de liaison à GP Ib/IX plaquettaire
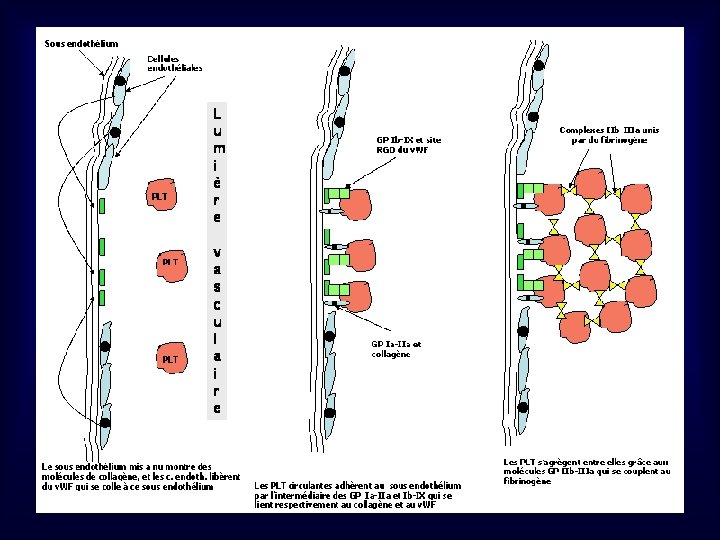

3. ACTIVATION PLAQUETTAIRE Changement de forme Libération granulaire Activation métabolique • Inducteurs de l’activation: collagène, ADP endothélial, traces de thrombine • Mécanismes biochimiques de l’activation plaquettaire: PLC Pi. P 2 DG + IP 3 PKC Mobilisation Ca 2+ Fusion des granules PL mb PLA 2 Acide arachidonique Cox PGG 2 Txs Calmoduline TXA 2 Changement forme Sécrétion granulaire vasoconstriction et agrégation plaquettaire

• Changement de forme: discoïdes initialement, PLT deviennent sphériques et volumineuses, et émettent des pseudopodes. • Relargage du contenu des granules plaquettaires: centralisation des granules et fusion avec les systèmes canaliculaires ouverts sur l’extérieur -granules denses : Ca 2+, ATP, ADP, adrénaline, sérotonine, -granules : fibrinogène, fact. V, v. WF, PDGF, fibronectine, TXA 2 -lysosomes: hydrolases, élastases, collagénases Ces substances, notamment ADP et TXA 2, stimulent le recrutement et l’activation de PLT voisines, qui subissent les mêmes modifications morphologiques et métaboliques • Remaniements mb : mécanisme flip-flop des phospholipides anioniques -constitution du « F 3 P » pour fixation Ca 2+ dépendante des facteurs de la Coagulation vit. K dépendant (II, VII, IX, X). -accessibilité de GPIIb/IIIa (capable aussi de fixer v. WF)

4. AGREGATION PLAQUETTAIRE • GP IIb/IIIa mb : récepteur au Fibrinogène permet la formation de ponts « fibrinogène » en présence de Ca 2+, entre PLT voisines • assurée par l’enchevêtrement des pseudopodes • dernier stade: fusion des mb PLT entre elles Rq: Dernière phase de l’hémostase après coagulation: Rétraction du caillot après la coagulation, les PLT interviennent encore par l’activité de leur protéines contractiles, pour contracter l’agrégat et exsuder le sérum retenu

5. REGULATION ACTIVATION PLAQUETTAIRE • adhésion plaquettaire reste localisée car elle n’a lieu uniquement que sur sous-endothélium lésé • TXA 2 rapidement inactivé en TXB 2 inactif • Cellules endothéliales produisent à partir de l’acide arachidonique libéré, de la Prostacycline PGI 2: vasodilatateur et anti-agrégant plaquettaire La PGI 2 favorise l’activation de l’adénylate cyclase ATP AMPc L’augmentation d’AMPc favorise le stockage du Ca 2+ dans les structures tubulaires denses des PLT, donc diminue l’agrégation plaquettaire

6. EXPLORATION DE L ’HEMOSTASE PRIMAIRE • Mesure du Temps de Saignement – TS -: explore l’hémostase primaire dans son ensemble, vaisseau + plaquettes + protéines. Temps nécessaire à l’arrêt saignement provoqué par une petite coupure cutanée au niveau des vaisseaux superficiels. -Méthode d’Ivy: avant-bras, 3 points de piqûre ou incision 1 cm, sous pression 50 mm. Hg. TS < 10 min. -Méthode de Duke (moins reproductible, moins sensible): lobe de l’oreille, incision. TS < 5 min. L’allongement du TS: anomalie quantitative ou qualitative des plaquettes, anomalies vasculaires, anomalies des facteurs plasmatiques • Numération Plaquettaire: Risque Hémorragique PLT < 50 000/mm 3 mais rarement hémorragie grave sauf si PLT < 20 000 /mm 3 en association avec anomalie hémostase ou lésion viscérale

• Mesure du Temps d’Occlusion sur PFA-100 Dade-Behring Analyseur reproduisant l’environnement d’un vaisseau lésé: -réservoir de sang total citraté -mb nitrocellulose enduite : Collagène + ADP ou +Adrénaline -capillaire 200µm abouché à la membrane percée d’un orifice 150µm, qui simule la brèche vasculaire Méthode: -Passage du sang total par le capillaire (800 à 1000µl) et par l’orifice de la membrane (sous aspiration constante créant des forces de cisaillement représentant l’hémodynamique d’une artériole). -Adhésion et agrégation des PLT sur la membrane et entre elles, réduction du flux sanguin jusqu’à obstruction orifice et l’arrêt du flux. -Mouvement du sang à travers l’orifice mesuré par microprocesseur Mesure d’un Temps d’Occlusion: capacité fonctionnelle des plaquettes à constituer un agrégat, méthode alternative à la mesure du TS in vivo Plus sensible et reproductible que TS, permet un bon dépistage du déficit en v. WF, et est plus sensible que TS à la baisse de l’agrégation induite par un traitement à l’aspirine

Le tandem collagène – ADP [valeurs N = 70 – 110 sec (m = 90)] détecte mieux que le TS, les déficits en v. WF, cause la plus fréquente d’anomalie constitutionnelle de l’hémostase (allongement pratiquement constant du test collagène – ADP) Le tandem collagène – adrénaline [valeurs N = 100 – 170 sec (m = 130)] dépiste bien le traitement à l’aspirine. Il est ici aussi meilleur que le TS (par contre le PFA – 100 détecte mal les prises de clopidogrel ou de ticlid) NB : quelques restrictions d’emploi à ce test sur appareil : Hte < 25% ou Hb < 6 g/dl, PLT < 70 G/L, prélèvement > 4 heures

Tests complémentaires • Adhésivité PLT: sur billes de verre ou sur sous-endothélium animal, peu réalisé • Agrégation PLT in vitro: turbidimétrie à 37°C sur PRP, ou sur PLT lavées en suspension agitée, en présence d’agents inducteurs tels que ADP, Acide arachidonique, collagène, thrombine, ristocétine. La transmission lumineuse de la suspension augmente lorsque se forment les agrégats. • Activation PLT: dosage substances libérées sous stimulation: ADP, ATP, thromboglobuline, PF 4, TXB 2… • Dosage Facteur von Willebrand: - dosage immunologique avec Ac spécifiques v. WFAg : ELISA, Latex, Laurell - dosage fonctionnel v. WFRCo (activité cofacteur de la ristocétine) mesure turbidimétrique de l’agrégation de PLT normales lavées en présence du plasma du patient et d’un inducteur de l’agrégation, la ristocétine • Dosage Fibrinogène: chronométrique ou immunologique

• Recherche des Glyco. Protéines membranaires des PLT: par cytométrie de flux, à l’aide d’Ac spécifiques marqués par des fluorochromes • Mesure de la résistance capillaire : exploration qualité des vaisseaux résistance des parois des capillaires cutanés à une dépression d’intensité variable: dépression 10 cm Hg par ventouse au pli du coude pendant 5 min. , dénombrement du nombre de pétéchies. Fragilité capillaire si nbre de pétéchies > 5


7. PATHOLOGIES DE L’HEMOSTASE PRIMAIRE TS allongé Num. PLT Patient : Prise médicamenteuse ? Dosage Fibrinogène Dosages v. WF Tests Fonctionnels PLT Résistance capillaire • Atteintes vasculaires: Fragilité capillaire • Thrombopénies • Thrombopathies • Maladie de Willebrand syndromes hémorragiques cutanés et muqueux • Thrombocytoses: associées SMP, asplénies, hémorragies, hémolyses, anémies microcytaires hypochromes risque thrombotique

7. 1 Maladie de WILLEBRAND La plus fréquente des maladies hémorragiques constitutionnelles: incidence 10 -15/105 habitants, Prévalence 1 à 2 % -Transmission autosomale (chr 12) dominante le + svt, sauf dans la forme la plus sévère qui est de transmission récessive -Syndrome hémorragique cutanéo-muqueux (ecchymoses, épistaxis, hémorragies viscérales…) d’intensité extrêmement variable selon le niveau de déficit, selon l’âge, … -3 grands types de Déficits en v. WF: *type 1: quantitatif partiel, le + fréquent 70%-80% des patients *type 3: quantitatif quasi-total, le + grave, 3% des patients *types 2: qualitatifs, 4 variants, 21 sous-types 2 a: absence de multimères, diminution affinité pour PLT 2 b: multimères absents dans plasma, mais présents dans PLT, hyperaffinité pour GPIb 2 M: diminution affinité pour PLT 2 N: anomalie de liaison au VIII Rq: v. WF transporteur VIII plasmatique: si déficit primaire en v. WF, très souvent déficit secondaire en VIII

2 maladies hémorragiques constitutionnelles de l’Hémostase les plus fréquentes: symptômes cliniques peu différents Maladie de Willebrand type 1 ou 3 Hémophilie A TS Allongé (ou Normal) Normal TQ-TP% Normal TCA Allongé VIII Diminué ou Nul v. WFAg Diminué ou Nul Normal v. WFRCo Diminué ou Nul Normal
 A")
Diagnostics différentiels types Willebrand Type 1 Types 2 Type 3 A (ou N) A ou N A D D ou N D v. WFAg D D ou N Nul v. WFRCo D Très D (2 a, 2 b) ou N Nul Agrégation PLT à Ristocétine D D (2 a), ou A (2 b) ou N Nulle Multimères plasma N Anx (2 a, 2 b) ou N - Multimères PLT N Anx (2 ae 1) ou N - Fixation GPIb N D (2 a, 2 M) ou A (2 b) ou N - Fixation au VIII N N ou D à Nulle (2 N) - TS VIII (A: allongé, N: normal, D: diminué, Anx: anormaux)

• Traitement Willebrand - augmentation libération v. WF par cellules endothéliales pour type 1 et certains types 2 (sauf 2 b) par DDAVP (analogue de la vasopressine) - substitution par injection de concentrés plasmatiques VWF viro-inactivés • Prophylaxie des hémorragies: contre-indication de l’aspirine, ou anti-agrégants PLT, et de certains anti-inflammatoires.
 -constitutionnel héréditaire: Maladie de Rendu-Osler -infectieux:")
7. 2 Atteintes VASCULAIRES: purpuras (pétéchies hémorragiques cutanés) -constitutionnel héréditaire: Maladie de Rendu-Osler -infectieux: Méningocoque, virus, parasites… -immuno-allergique médicamenteux: pénicilline, aspirine, sulfamides -rhumatoïde ou associé à d’autres MAI
 -Origines centrales: aplasie,")
7. 3 THROMBOPENIES (attention fausses thrombopénies dues à EDTA, prélèvement Citrate) -Origines centrales: aplasie, hémopathies malignes, métastases, virus, intox. alcoolique aiguë, benzène médicaments (chimiothérapie, antiviraux, radiothérapie, …) -Périphériques: * par consommation: CIVD et Purpura fulminans à Méningocoque ~ CIVD: Coagulation Intra. Vasculaire Disséminée ~Microangiopathies (Purpura Thrombotique Thrombopénique, Syndrome Hémolytique et Urémique, HELLP syndrome) ~iatrogène (Circ. Extra-Corporelle. , transfusion massive…) * par anomalie de répartition: ~hypersplénisme (le plus svt associé à cirrhose alcoolique en France)

* par destruction immunologique : Ac anti-mb PLT ~ Purpura Thrombopénique Auto-Immun –PTAI- : -isolé, considéré comme idiopathique (PTI) ou -associé à MAI, Hémopathie maligne, virus ~ immuno-allergique: HEPARINE, quinine, digitaline, sulfamides, sels d’or, methyldopa… ~ allo-immunisation: (groupes plaquettaires) thrompopénies néonatales (allo-Ac anti-PLT d’origine maternelle) et thrombopénies post-transfusionnelles Ac le + fréquent : anti-HPA 1 a

7. 4 THROMBOPATHIES : anomalies qualitatives fonctionnelles * acquises les + fréquentes: - associées aux SMP, SMD, … - médicamenteuses: ASPIRINE, PENICILLINE, Ticlopidine, Dipyridamole, … * constitutionnelles: rares (1/10 Millions) - Dystrophie de Bernard-Soulier: déficit GP Ib-IX - Thrombasthénie de Glanzmann: déficit GP IIb-IIIa - Maladie du pool vide: diminution contenu granules denses - Syndrome des Plaquettes grises: diminution contenu granules - Déficit en Cyclo-oxygénase Perturbation des tests d’agrégation in vitro des PLT des patients aux différents agents inducteurs

Réponses plaquettaires typiques à différents agents inducteurs en PRP

Tableau synthétique des principales anomalies des tests d’agrégation plaquettaire agoniste Glanzma nn (GP IIb. IIIa) Bernard Soulier (GP Ib) Pseudo. Willebra nd Déficit en granule s denses Déficit granules (PLT grises) Syndro me "Aspirinlike" Réversibl e (abs. 2 ième vague) Diminuée ou nulle ADP nulle N N Diminué e et réversibl e N Collagèn e nulle N N Presque nulle diminuée Ac. Arachidonique nulle N N Diminué eparfois N N Ristocéti ne Présente mais diminuée Nulle Hypoagr ég. à faible dose N adrénali ne nulle N N N Anomal ie récepte ur ADP "Ticlopi -dinlike" Diminué e Déficit GP Ia. IIa N Diminué e Nulle N ou dim. N N N Abs. 2 ième vague N N

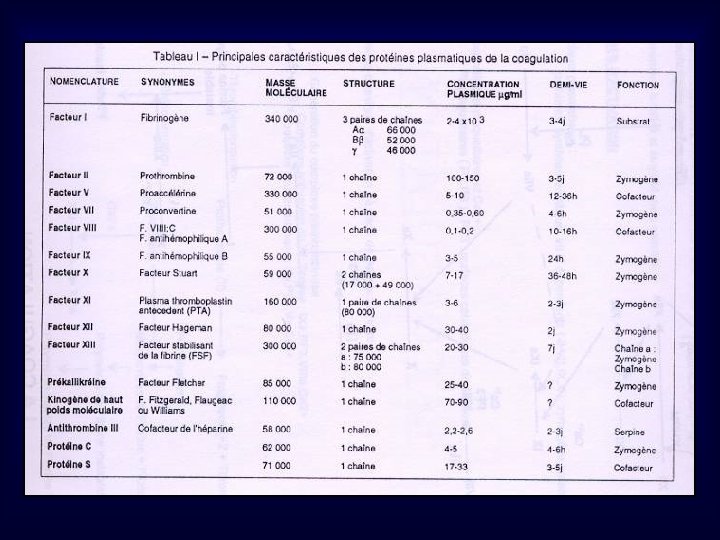
LA COAGULATION Deuxième étape de l’hémostase, conduisant à la formation de fibrine, permettant de consolider l’agrégat plaquettaire Implique des facteurs plasmatiques, des protéines de l’inflammation, des phospholipides, du Ca 2+ 1 -Propriétés des Facteurs de la Coagulation -synthèse majoritairement hépatique -désignés par chiffres romains, et suffixe « a » sous forme activée -Facteurs Vitamine K dépendants nécessite l’intervention de la vit. K, pour une synthèse sous forme fonctionnelle: II, VII, IX, X Modification post-traductionnelle: g carboxylation de résidus Acide Glutamique (GLU) de la chaîne protéique, par une g carboxylase utilisant la vit. K comme cofacteur enzymatique
 formés, situés dans la région Nterminale, confèrent à ces facteurs la")
Les g-carboxyglutamates (GLA) formés, situés dans la région Nterminale, confèrent à ces facteurs la propriétés de fixer le Ca 2+ et de se lier au phospholipides anioniques des mb plaquettaires et cellulaires L’absence de vit. K ou son inhibition provoque la production de facteurs II, VII, IX, X non fonctionnels appelés PIVKA (Protein Induced by Vitamine K Absence or Antagonist)

-facteurs du système des kinines inflammatoires: Prékallicréine, Kininogène de Haut Poids Moléculaire KHPM -Facteur Tissulaire appelé aussi Thromboplastine tissulaire: facteur glycoprotéique comportant une partie phospholipidique, synthétisé par de nbx types cellulaires, libéré lors de lésion vasculaire -Du point de vue fonctionnel, les facteurs de la coagulation, sont, soit: -des zymogènes: proenzymes capables d’acquérir une activité enzymatique protéolytique (sérine-protéases) par coupure -des cofacteurs de réaction: augmentent la vitesse d’activation du substrat par enzyme, en formant des complexes Enz. Cofacteur-Substrat -un substrat terminal: le fibrinogène

2 -Phospholipides et Calcium • Phospholipides: -surface catalytique pour activation des facteurs de la coagulation, -permet de concentrer et de faciliter les interactions entre protéines, -permet de localiser l’action de la coagulation uniquement au niveau de l’agrégat plaquettaire, donc de la brêche vasculaire -PF 3: facteur 3 plaquettaire -partie phospholipidique du Facteur Tissulaire • Calcium: indispensable à certaines activités enzymatiques, et permet fixation des Facteurs Vit. K dépendants sur phospholipides Prélèvement sur Anticoagulants (Chélateurs du Ca 2+ ) pour obtenir Plasma: EDTA potassique, Citrate de Na+

3 -Déroulement de la coagulation 3 étapes successives: -la génération d’un complexe prothrombinase: 2 voies possibles -la thrombinoformation -la fibrinoformation • Génération d’un complexe prothrombinase a)voie intrinsèque ou endogène: activation de facteurs de la phase contact PK, KHPM, XII, XI In vivo: surface contact activatrice électronégative du collagène sous-endothélial, et la protéolyse de la PK sur les ¢ endothéliales semblent initier la voie In vitro: surface contact activatrice électronégative type verre, kaolin, silice, provoque changement conformationnel XII -activations successives du XII, XI, IX -formation d’un complexe Tenase (IXa, VIIIa)+ Ca 2+ + PF 3: activation X
voie extrinsèque ou exogène: la lésion vasculaire s’accompagne d’une libération de Facteur Tissulaire -formation")
b)voie extrinsèque ou exogène: la lésion vasculaire s’accompagne d’une libération de Facteur Tissulaire -formation d’un complexe VII- Ca 2+ -FT, où le VII s’active en VIIa, puis activation du X • Thrombinoformation -formation complexe prothrombinase : association Xa + Ca 2+ + Va, liés à surface phospholipidique (mb plaquette PF 3, ou FT) -clivages de la Prothrombine II en Thrombine IIa -Thrombine: enzyme puissante, libre, plusieurs rôles -fibrinoformation -activation facteur V, VIII, XIII -participe à l’activation et agrégation plaquettaire dans Hémostase primaire -participe à l’activation de Protéine C (inhibiteur physiologique)

• Fibrinoformation : Fibrinogène: 6 chaînes polypeptidiques A Bb gg, identiques 2 à 2 -clivage extrémités N-Terminales des chaînes A Bb : formation de monomères de fibrine -polymérisation des monomères de fibrine par liaisons H: formation de fibrine instable et soluble -stabilisation de la fibrine par liaisons covalentes de transamidation grâce au XIIIa, entre les domaines appelés D (région Cterminales) des monomères: formation de fibrine insoluble

Voie endogène PréKallicréine Surface électronégative collagène sousendothélial Voie exogène KHPM XII Facteur Tissulaire XIIa KHPM XI FT-VIIa XIa VII + Ca 2+ PL+Ca 2+ IX VIII IXa VIIIa X Ca 2+ + PF 3 Xa Ca 2+ + PL Prothrombine II X Va V XIII Thrombine IIa Fibrinogène Fibrine soluble Voie exogène: voie principale in vivo Voie endogène: voie de consolidation XIIIa Fibrine
")
4 -Les inhibiteurs physiologiques de la coagulation 4 -1 Les inhibiteurs des Sérine-protéases (SERPINS) Inhibiteurs protéiques formant des complexes équimoléculaires stables, inhibant site actif (comportant résidus sérine) des facteurs activés • Anti. Thrombine III (ATIII) -glycoprotéine plasmatique, synthétisée par le foie -inhibiteur majoritairement de Thrombine et Xa -inhibiteur modéré de IXa, XIIa, et Kallicréine -action lente, mais fixation d’Héparine sur ATIII, augmente vitesse d’inhibition (x 2000), d’où l’action anticoagulante de l’Héparine -In vivo, l’héparane-sulfate (glycosaminoglycanne) de la paroi vasculaire, joue le rôle de l’héparine ATIII représente 77% de l’activité d’inhibition de la thrombine dans le plasma • Cofacteur 2 de l’Héparine (HCII): inhibition spécifique de la Thrombine, potentialisée in vivo par le dermatane-sulfate, et l’héparine

4 -2 Autres Inhibiteurs • 2 -Macroglobuline, 1 -antitrypsine, C 1 -Inhibiteur: inhibiteurs modérés Thrombine IIa, Kallicréine et XIIa XIa, Xa XIIa, Kallicréine • Système de la Protéine C / Protéine S: -Protéine C: zymogène, synthèse hépatique vit. K dépendante -Protéine S: cofacteur, synthèse hépatique, endothéliale, et Mk -Thrombomoduline: protéine mb récepteur de la thrombine sur la ¢ endothéliale Protéine S + Ca 2+ Inactivation Va Protéine Ca Inactivation VIIIa Protéine C Cellules endothéliales Thrombomoduline

Système PC / PS : 3 actions inhibitrices de la coagulation -protéolyse du Va et VIIIa -piège la Thrombine 1 action activatrice de la fibrinolyse: -inactivation de l’inhibiteur (PAI) du t-PA • Tissue Factor Pathway Inhibitor (TFPI) -synthèse endothéliale et Mk -complexe le Xa, et devient inhibiteur du complexe FT-VIIa

5 -L’exploration biologique in vitro de la coagulation 5. 1 -Temps de coagulation sang total in vitro: obsolète 5. 2 -Temps de Quick TQ -exploration de la fonctionnalité de l’ensemble des facteurs de la voie exogène et de la voie commune: Fibrinogène, II, V, VII, X -Temps de coagulation à 37°C d’un plasma citraté, déplaquetté, en présence de thromboplastine tissulaire et de Ca 2+ -Expressions du résultat: TQ en sec. , Taux de Prothrombine TP en %, ou INR (International Normalized Ratio) dans le cas de traitement AVK 5. 3 -Thrombotest d’Owren: peu utilisé, identique au TQ, mais pas influencé par V et Fibrinogène
 TCA -exploration fonctionnelle de l’ensemble")
5. 4 -Temps de Céphaline + Activateur (ou Activée) TCA -exploration fonctionnelle de l’ensemble des facteurs de la voie endogène et de la voie commune: Fibrinogène, II, V, VIII, IX, X, XII, et PK, KHPM -Temps de coagulation à 37°C d’un plasma citraté, déplaquetté, en présence d’un substitut phospholipidique plaquettaire, la céphaline, de Ca 2+, et d’un activateur des facteurs de la phase contact, silice, kaolin… -expressions du résultat: TCA en sec. , ou Ratio TCA patient / témoin 5. 5 -Temps de Thrombine TT -exploration de la fonctionnalité de l’étape de fibrinoformation (sauf XIII) -temps de coagulation à 37°C d’un plasma citraté, déplaquetté, en présence de Ca 2+, et d’une quantité déterminée de thrombine -expression TT en sec.
 du Fibrinogène")
5. 7 -Dosage du Fibrinogène • Dosage chronométrique (dit de von Clauss) du Fibrinogène -dosage indirect, car mesure d’un temps d’apparition de fibrine par transformation de la totalité du fibrinogène présent -temps de coagulation à 37°C d’un plasma dilué, citraté, déplaquetté, en présence de thrombine concentrée, est inversement proportionnel à la quantité de fibrinogène -dosage quantitatif et qualitatif -expression en g/L • Dosage immunologique du fibrinogène -en général par immunonéphélémétrie -dosage quantitatif, pas fonctionnel

5. 8 -Dosage chronométriques de facteurs isolés de la coagulation à l’aide de plasmas déficients -Plasmas réactifs déficients en un seul facteur -Temps de coagulation (TCA ou TQ en fonction du facteur à doser) d’un plasma déficitaire est fonction de la quantité du facteur apporté par le plasma à tester -Résultats exprimés en % de facteur par rapport à des plasmas normaux servant d’étalons 5. 9 -Dosage colorimétriques ou amidolytiques -utilisation de substrats oligopeptidiques chromogènes: groupement paranitroaniline libère amine p. NA par coupure enzymatique -dosage d’activité enzymatique: concentration enzyme (facteur) est proportionnelle à la vitesse d’hydrolyse, mesurée par la variation absorbance (dosage pt final, 2 pts, cinétique…) - dosage isolé de chaque facteur, ou des inhibiteurs physiologiques 5. 10 -Dosage par techniques immunologiques: Immunonéphélémétrie, ELISA -dosage isolé de chaque facteur de la coagulation, ou d’inhibiteurs f

LA FIBRINOLYSE Processus physiologique permettant destruction du caillot de fibrinoplaquettaire qui se déclenche dès que la cicatrisation de la brèche vasculaire est amorcée. 1 -Mécanisme Fibrine Produits de Dégradation de la Fibrine PDF PLASMINE Plasminogène -Plasminogène: glycoprotéine plasmatique, synthétisée par foie -Activation plasminogène libère plasmine, qui reste localisée au niveau de la fibrine -dégradation progressive de la fibrine en PDF, de plus en plus courts -tous les PDFibrine contiennent structure domaines D-D appelée D-Dimères (car les liaisons covalentes entre monomères de fibrine ne sont pas rompues par la plasmine) -existence de D-Dimères: preuve de la formation de fibrine stabilisée donc d’une coagulation, puis de sa lyse par plasmine

2 - Activation du plasminogène Plasmine t-PA Urokinase Kallicréine • t-PA: activateur tissulaire du plasminogène -synthèse endothéliale vasculaire continue, mais libération massive si anoxie tissulaire, traumatisme endothélial, vasoconstriction, stase sanguine, ischémie… -t-PA libre, fixé par fibrine, et forme complexe t-PA-Fibrine. Plasminogène • Urokinase: pro-urokinase synthétisée par ¢ rénales, plasmatique, et activée par Kallicréine • Kallicréine: facteur système contact de la coagulation • Activateur non physiologique: la streptokinase (exo-enzyme Sb. HA)

3 -Autres actions de la plasmine Dans certaines conditions d’activation, (excès de formation par rapport aux capacités d’inhibition, lors de libération massive de t-PA): -action fibrinogènolytique: la plasmine est capable de dégrader du fibrinogène en Produits de Dégradation du Fibrinogène, PDFib, qui ne contiennent pas de structures D-Dimères Plasmine Fibrinogène Fragments X, Y, D, E -destruction du VIII, V, XIIIa Rq 1: les PDF ont une action inhibitrice sur la coagulation (action antithrombine, inhibition de la polymérisation de fibrine, anti-agrégation plaquettaire) Rq 2: activation de la fibrinolyse et fibrinogènolyse d’origine leucocytaire (PN), par élastase, et cathepsine

4 -Régulation de la fibrinolyse • Inhibiteurs de l’activation du plasminogène PAI (Plasminogen Activation Inhibitors) -PAI 1: synthèse par endothélium, PLT, foie, inhibition du t-PA, et urokinase, variations circadiennes (max matin, min soir), augmentation avec âge, grossesse, état inflammatoire -PAI 2: synthèse placentaire, inhibition urokinase • Inhibiteurs de la plasmine - 2 -antiplasmine: lie la plasmine libre, et le plasminogène - 2 -Macroglobuline, 1 -Anti. Trypsine, C 1 -Inhibiteur: inhibiteurs modérés

5 -Exploration biologique in vitro de la fibrinolyse recherche les indices d’une hyper-fibrinolyse 5. 1 -Tests directs • Temps de lyse d’un caillot de sang dilué: obsolète, trop long • Temps de lyse du caillot des « euglobulines » (Test de von Kaulla) « euglobulines » ? ? : activateurs de la fibrinolyse, fibrinogène, plasminogène -précipitation des euglobulines par dilution plasma et acidification -re-solubilisation en tampon -activation de la coagulation par addition de Ca. Cl 2 et thrombine -mesure du temps de lyse du caillot de fibrine formé: entre 3 et 6 H -si < 2 H: témoin d’une hyper-fibrinolyse • Dosage Plasminogène par technique amidolytique • Dosage du t-PA , urokinase, par ELISA
 -tech.")
5. 2 -Test indirects • Recherche et Dosage des PDF (Fibrine et Fibrinogène) -tech. Immuno. par agglutination latex sensibilisé -PDF sériques > 10 mg/L: témoin hyper-fibrino ou fibrigèno-lyse • Dosage des D-Dimères -tech. ELISA et tech. Immunoturbidimétrique -D-Dimères sont spécifiques de l’activité de fibrinolyse, donc leur concentration augmentent dans sérum après thrombose, mais aussi lors de circonstances variées (âge, grossesse, cancers, infections, inflammation, post-opératoire…) -utilisation du dosage: dosage à valeur prédictive négative, proche de 100%, d’une thrombose si D-Dimères sériques < 500 µg/L, exclusion du diagnostic d’un événement thrombotique chez un patient Permet d’éviter investigations (angiographies, scintigraphies) difficiles, invasives et coûteuses
- Slides: 52