COSMORS The basic principle and the wider application

COSMO-RS: The basic principle and the wider application range Andreas Klamt COSMOlogic Gmb. H&Co. KG Leverkusen, Germany & Inst. of Physical and Theor. Chemistry, Univ. of Regensburg

The dimensionality of the solvent space Scatchard and Hildebrand introduced solubility parameters as a measure of solute-solvent compatibility. But compressed the world of solutes and solvents to one dimension! Charles Hansen realized that the world of solutes and solvents is not 1 -dimensional. 50 years ago he introduced the 3 D-space of Hansen solubility parameters by splitting the cohesion energy into dispersive, polar and hydrogen bonding contributions. Since mankind is so much used to think in 3 dimensions, the HSP concept of measuring distances in this 3 -dimensional space became very popular. And despite of its rather empirical basis, it has become very sucessful in explaning many solubility phenomena an making useful predictions!!! Congratulations for this achivement to Charles Hansen and to the HSP team!

But are 3 dimensions enough? Most likely not: At least hydrogen bonding cries for a splitting into donor and acceptor strength. MOSCED (Modified Separation of Cohesive Energy Density, Thomas. E. R. ; Eckert C. A. (1984)) has ~4 dimensions (6 parameters) But one has to give up the simple distance scheme and who can think in 6 dimensions? Mike Abraham (University College London) introduced a 5 -dimensional scheme of solute parameters and solvent coefficients, but only for infinite dilution, not for mixtures. And altogether he needs 10 parameters for each compound. None of these concepts was as successful as HSP!

Maybe we need a molecular structure view? Full quantum chemistry for all molecules of a liquid is still utopic! force field models (with lots of approximations and thousands of parameters) But either molecular dynamics or Monte-Carlo statistics is needed: After weeks of computations on huge computers you yield free energies which have error bars of ~ 1 kcal/mol As you can clearly see, you see nothing! mixture of ethanol and heptafluoropropane Grottel et al. , Computer Graphics Forum 29(3): 953 -962 · June 2010

COSMO-RS Chemical Structure Phase Diagrams provides an intermediate level of complexity: 3 D-molecules with QM-info solubility Quantum Chemical Calculation in a conductor provides surface polarity Equilibrium data: activity coefficients vapor pressure, solubility, partition coefficients -potential of mixture -profiles of compounds ideally screened molecule energy + screening charge distribution on surface Fast Statistical Thermodynamics -profile of mixture DFT/COSMOtherm
")
Step 1: Do DFT quantum chemistry in the presence of an embedding conductor (COSMO) to get polarization charge density on the molecular surface. con du cto electron density conduc tor r The conductor can be taken into account during the quantum chemical calculation using the Conductor-like Screening Model (COSMO, Klamt, Schüürmann, 1993) polarization charge density = polarization charge / area

Step 3: Quantify contact interactions of molecules swimming in a conductor using and ‘. ‘ no interactions of molecules in conductor!!! We only get an energy change DE, if the two cavities contact each other. Basic idea of COSMO-RS: Quantify interaction energies as local interactions of COSMO polarization charge densities and ‘ ‘ DEcontact = E( , ‘)

Step 2: Quantify contact interactions of molecules swimming in a conductor using and ‘. ‘ Interaction energy of individual contacts: Step 3: consider a dense liquid In a dense liquid all surface pieces are bound in surface pairs, and the total interaction energy can be expressed as a sum of surface interactions Eint( , ‘). But for liquid phase properties we need free energies, i. e. contact probabilities of all possible contacts!

Step 4: Split the molecular surface into contact segments and make a histogram -profiles. For an efficient statistical thermodynamics we reduce the ensemble of molecules to an ensemble of pair-wise interacting surface segments. For handling this we need histograms of surface polarity. Screening charge distribution on molecular surface reduces to " -profile"

Step 4: Split the molecular surfaces into contact segments and make histograms -profiles. Screening charge distribution on molecular surface reduces to " -profile"

Qualitative thermodynamics based on -profiles Why does it get warm when you mix acetone and? Because their -profiles are almost complementary! By the way, the activity coefficients of acetone in chloroform and vice versa are smaller than one, i. e. they are more happy in eachother than in themselves. HSP theory cannot handle such cases!

Statistical Thermodynamics • Replace ensemble of interacting molecules by an ensemble S of interacting pairs of surface segments • Ensemble S is fully characterized by its -profile p. S( ) of mixtures is additive! -> no problem with mixtures! • Chemical potential of a surface segment with charge density is exactly(!) described by: -potential: affinity of solvent for specific polarity chemical potential of solute X in S: combinatorial contribution: solvent size effects activity coefficients arbitrary liquid-liquid equilibria chemical potential of solute X in the gasphase: vapor pressures

-profiles and -potentials of representative liquids hydrophobicity affinity for HB-donors affinity for HB-acceptors
 COSMO-RS knows the")
2 fold For flexible molecules there are multiple local minima (conformations) COSMO-RS knows the internal energy (from DFT) and the individual free energy from central COSMO-RS eq. At every temperature and composition COSMO-RS can calculate the total free energy of the compound (from the partition function) and the exp. values of the all properties.

Chemical Structure Phase Diagrams Flow Chart of COSMO-RS Quantum Chemical Calculation with COSMO (full optimization) Equilibrium data: activity coefficients vapor pressure, solubility, partition coefficients -potential of mixture -profiles of compounds ideally screened molecule energy + screening charge distribution on surface Database of COSMO-files (incl. all common solvents) other compounds DFT/COSMO Fast Statistical Thermodynamics -profile of mixture COSMOtherm
 prediction: Accuracy (kcal/mol)")
COSMOtherm currently is the most accurate tool for solvation energy (DGsolv) prediction: Accuracy (kcal/mol) on 2343 data (Minnesota DB) COSMOtherm 0. 48 (latest COSMOtherm: 0. 40) SM 8 0. 52 (fitted on this data set!) PCM ~ 0. 9 (only 3 solvents) A. Klamt, B. Mennucci, J. Tomasi, V. Barone, C. Curutchet, M. Orozco and F. Javier Luque, Acc. Chem. Res. , 2009, 42 (4), pp 489 COSMO-RS is winner of all the 3 IFPSCs (Industrial Fluid Property Simulation Challenges; AICHE/NIST) which were on free energy of transfer (e. g. activity coefficients) and on which COSMO-RS was allowed to participate. Two benchmarks vs. Force-Field-MD show that the error on DGsolv is only about half of the error of force-field methods. Force Field Benchmark of Organic Liquids. 2. Gibbs Energy of Solvation Jin Zhang†‡, Badamkhatan Tuguldur§‡, and David van der Spoel J. Chem. Inf. Model. , 2015, 55 (6), pp 1192– 1201 (see also the erratum): ~230 DGsolv RMSD: MD 3. 7 k. J/mol, ADF-COSMO-RS 2. 3 k. J/mol, COSMOtherm 1. 7 k. J/mol. --------------Current Status of the AMOEBA Polarizable Force Field Ponder et al. , J Phys Chem B. 2010; 114(8): 2549: 22 DGsolv RMSD: AMOEBA 2. 7 k. J/mol, COSMOtherm 1. 6 k. J/mol SAMPL blind challenges for prediction of DGsolv/transfer SAMPL 2009: 45 entries from molecular dynamics, Monte Carlo, Continuum Solvation Models, and other methods: COSMO-RS error is about 0. 5 kcal/mol smaller than that of the second best entry. J. Phys. Chem. B 2009, 113, 4508– 4510 ----------March 2016: SAMPL 5: COSMOlogic clearly wins the SAMPL 5 blind challenge on alkane-water log. Ds of 53 demanding drugs and is better than the experimental values. J Comput Aided Mol Des (2016). doi: 10. 1007/s 10822 -016 -9927 -y

Winner of the Applications to Phase Diagrams and Azeotropes 1 st, 5 th, 6 th IFPSC (AICHE/NIST) miscibility gap

COSMOtherm prediction of drug solubility in diverse solvents (blind test performed with Merck&Co. , Inc. , Rahway, NJ, USA) all predictions are relative to ethanol solvents: triethylamine heptane Water 1 -Propanol 2 -Propanol DMF Ethyl Acetate Methanol Heptane Toluene Chlorobenzene Acetone Ethanol Acetonitrile Triethylamine Butanol

Example Absolute solvent screening with estimated DGfus All data are simulated / measured at 20°C Yellow points indicate alternative experimental measurements, the experimental range is additionally visualized by black lines. Data and DGfus are extracted from Lapkin A. , Peters M. , Greiner L. , Chemat S. , Leonhard K. , Liauw M. , Leitner M. , Screening of new solvents for artemisinin extraction process using abinitio methodology, Green Chem. , 2010, DOI: 10. 1039/b 922001 a Artemisinin:

Ionic systems and electrolyte applications: II Applications of COSMOtherm to Ionic Liquids exp. : J. G. Huddleston, University of Alabama Without any modification, COSMOtherm appears to work well for Ionic Liquids
: similar rms")
Ionic systems and electrolyte applications: III latest results for bases (p. Kb): similar rms formicacid aceticacid chloroaceticacid dichloroaceticacid 0 trichloroaceticacid n-pentanoicacid 2, 2 -dimethylpropanoicacid benzoicacid oxalicacid 0 maleicacid 3 fumaricacid carbonicacid 0 phenol pentachlorophenol ethanol 2, 2, 2 -trichloroethanol hypochlorousacid hypobromousacid hypoiodousacid nitrousacid sulfurousacid phosphoricacid 2 boricacid 5 -fluorouracil 5 -nitrouracil cis-5 -formyluracil thymine trans-5 -formyluracil Uracil and others

Ionic systems and electrolyte applications: IV Salt Effect on Activity Coefficients and VLEs exp. data from M. Topphoff, J. Gmehling, private communication

Probing Carboxylate Gibbs Transfer Energies via Liquid | Liquid Transfer at Triple Phase Boundary Electrodes: Ion. Transfer Voltammetry versus COSMO-RS Predictions Stuart M. Mac. Donald, Marcin Opallo, Andreas Klamt, Frank Eckert, and Frank Marken Submitted to PCCP
 localisation of")
Free Energies relevant for Reactions DGactivation Þ kinetic constant transition state (TS) localisation of transition state often complicated: In this work gas-phase TS have been localised using techniques provided in Gaussian 98 (DFT: B 3 LYP, 6 -31 G*) after that: single-point DFT/COSMO with TURBOMOLE (BP 91 -TZVP) DFT is not reliable for TS energies but the solvent shifts should be reliable. sum of educts sum of products DGreact Þequilibrium constant Calculation of the solvent dependence of DGreact is straightforward with COSMO-RS. Successful applications have been reported by industrial users (Dr. Franke, Degussa AG; Dr. Lohrenz, Bayer AG)

Accurate Prediction of Radical Propagation Rate Coefficients in Solution Polymerization Peter Deglmann, Imke Müller, Florian Becker presentated at Symposium for Theoretical Chemistry, Neuss, Germany, 9. 9. 2009 Conclusions • … • COSMO-RS describes (relative) solvent effects on reaction rate excellently (even in aqueous media) • …

Treatment of complex or chemically less well-defined matrices -consider a complicated liquid or amorphous matrix (liquid of complex or unknown composition, biological tissue, soap, adsorbent like active coal, polymer, . . . ) -> -potential cannot be calculated directly by COSMO-RS -> assume that S( ) can be composed of a set of basis functions fi( ) COSMO-RS says, that the solvent space is 5 -dimensional and that any log-partition coeff. Is a linear combination of a set of 5 -moments. M. Abraham (Impirial College) since long has a general solvation theory using 5 linear descriptors for log-partion coefficients! Now the chemical potential of a solute X in this matrix S is: The inter-correlation of the two sets of five linear solvation descriptors Has been investigated in: Zissimos, Abraham, Klamt, Eckert, J. Chem. Inf. Comput. Sci. , 42, 1320 -1331 (2002) The coefficients can now be derived from experimental (log. ) partition data by linear regression. => -moments are excellent QSAR-descriptors for general partition behaviour of molecules.
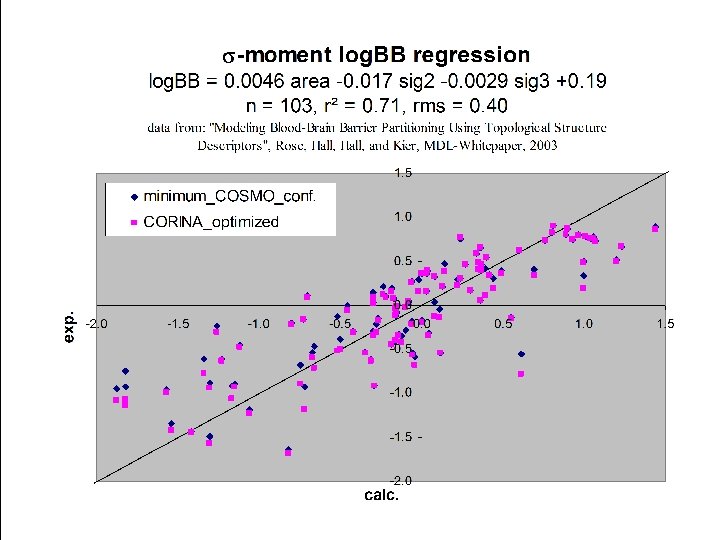

Prediction of Soil Sorption Journal of Environmental Toxicology and Chemistry, in print
![[Mehler, Peukert (TU München), Klamt; AICHE Journal. 48, 1093 -1099 (2002)]](http://slidetodoc.com/presentation_image_h/bf0a10a42c56693b654a2e4983cad766/image-29.jpg "[Mehler, Peukert (TU München), Klamt; AICHE Journal. 48, 1093 -1099 (2002)]")
[Mehler, Peukert (TU München), Klamt; AICHE Journal. 48, 1093 -1099 (2002)]
polymer Cut and cap DFT/COSMO e =")
Calculation of Polymer Properties General Approach amorphous (co)polymer Cut and cap DFT/COSMO e = infinity [ ] Construct -Profile from monomeric repeat unit [ ] [ ]
 log. Scalc. = log. SCOSMO +")
Solubility of Gases in Polymers (1 adjusted parameter/polymer) log. Scalc. = log. SCOSMO + const 3. 9 const 6. 0 (HDPE) (POP) 24 polymers exp. data: S. Pauly (Hoechst AG) Polymer Handbook 3 rd ed. , VI, 435 ff

Prediction of Flavor Sorption in Polymers by COSMOtherm

COSMOflat: Simulation of molecules at a flat interface Concept: - assume just a flat interface between two arbitrariry user-defined liquids, (typically one his hydrophobic and other is water) -for the free energy of a molecule at the interface use the local -potentials -construct a total partition sum - get the total free energy change of the solute at this interface from building the partition function over all positions and orientations - conformations can be taken into account, e. g. stretched vs. collapsed

COSMOmic: Simulation of molecules in micelles and membranes Concept: -define layers of membrane (shells of micelle) -get probability to find a certain atom of surfactant in each layer (e. g. from MD) -convert this into a -profile p( , r) for each layer r using the COSMO-file of the surfactant -use COSMOtherm to calculate µ( , r) considering each layer as a liquid mixture o o o -now calculate the chemical potential of a solute X in a certain position and orientation by summing the chemical potentials of its segments in the respective layer. -sample the chemical potentials all positions and orientations of X -construct a total partition sum and get the probability to find the solute in a certain depth and orientation. -also get the average volume expansion in each layer - get a kind of micelle or membrane-water partition coefficient The tool COSMOmic facilitates all the previous steps together with COSMOtherm Perspective: self-consistent treatment of new surfactants; CMC prediction
 Prediction of Phospholipid-Water Partition Coefficients")
COSMOmic: Simulation of molecules in micelles and membranes (2) Prediction of Phospholipid-Water Partition Coefficients of Ionic Organic Chemicals using the Mechanistic Model COSMOmic (J. Phys. Chem 2014 o o o COSMOmic: A Mechanistic Approach to the Calculation of Membrane-Water Partition Coefficients and Internal Distributions within Membranes and Micelles, J. Phys. Chem. B, 2008 Andreas Klamt, Uwe Huniar, Simon Spycher, and Jörg Keldenich|
 membrane MD-simulations*,")
Micelle and Interface Properties: COSMOmic Example: Free energy profiles through DMP (dimethylphthalate) membrane MD-simulations*, ** COSMOmic (5 minutes calc. time) * Simulation results of Daniele Bemporad and Jonathan W. Essex University of Southampton, UK and ** see also Claude Luttmann, J. Phys. Chem. B 2004, 108, 4875 ff

COSMOmic: limitations and drawbacks Drawback: - requires MD input Limitations: - infinite dilution of solute in micellar system - not applicable to the surfactant itself (e. g. cmc-prediction)
: Towards the self-consistent calculation of inhomogeneous systems Idea: Self-consistently calculate")
Self-consistent COSMOmic (COSMOmic. SC): Towards the self-consistent calculation of inhomogeneous systems Idea: Self-consistently calculate a pressure in each layer which brings the COSMOmic distribution of the system to homogeneous spacefilling interfacial tensions, CMC, curvature of interfaces, micelles with mixed composition, micelles with finite solute concentrations … First systems are converging but still a lot of work to be done!

Figure 1: COSMO polarization surface charge densities from Turbomole-cluster-BPSVP/COSMO calculations of 3 representative paracetamol faces (I-010, I-110, and I-011) Figure 2: COSMO polarization surface charge densities from Turbomole-cluster-BP-SVP/COSMO calculations of 3 representative paracetamol faces (I-010, I-110, and I-011)

COSMO-RS : Computation of Surface Free Energies: Results for Water Same trend in the different calculational approaches: A 1: DMOL//COSMO, A 2: DMOL//COSMO+COSMO-RS B: TURBOMOLE//BP//SVP//COSMO+COSMO-RS - Initial linear behaviour (but slope of 0. 5!) - Saturation at ~70 [m. N/m] - Speculation about the deviations: - Paracetamol-saturated water ? water saturated surfaces ? The most polar surface positions close to the droplet should already have a water molecule adsorbed thus blocking contact with the solvent, which leads to the saturation effect experimentally observed!
![Cocrystal-Screening with COSMOtherm § Initial tests gave better results than current state-of-the-art method [1]](http://slidetodoc.com/presentation_image_h/bf0a10a42c56693b654a2e4983cad766/image-41.jpg "Cocrystal-Screening with COSMOtherm § Initial tests gave better results than current state-of-the-art method [1]")
Cocrystal-Screening with COSMOtherm § Initial tests gave better results than current state-of-the-art method [1] (6 false positive results versus only 2 with COSMOtherm!) § Fast screening against large databases with harmless food ingredients (EAFUS, GRAS) within minutes with COSMOfrag technology. [1] Hunter et al. , Chem. Sci. 2011, 2, 883– 890.

Summary & Conclusions HSP is a great and easy to comprehend empirical scheme for structuring the space of solutes and solvents. Congratulations to 50 successful years of HSP! If want or need to lift the curtain slightly more, COSMO-RS provides a vivid molecular picture and a still reasonably simple, but more rigorous and broader applicable pathway to solubility predictions and general quantitative computational fluid phase thermodynamics.
- Slides: 42