CONNETTIVITI Lupus eritematoso sistemico Sindrome da anticorpi antifosfolipidi



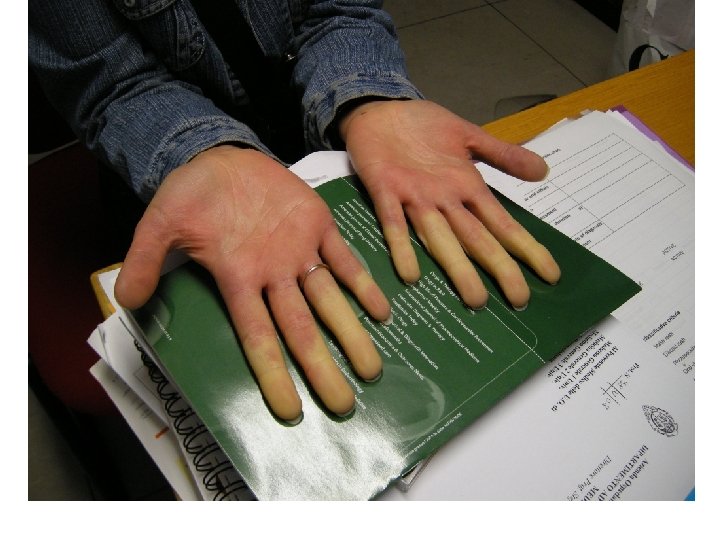

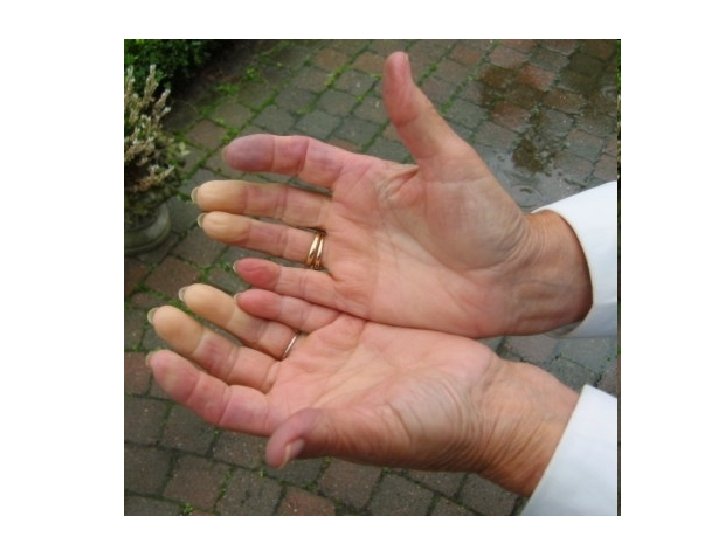
 • Ischemia acrale parossistica, scatenata dal freddo")
























 (%) Letteratura (%) Artralgie 82 53 -95")






























, anemia emolitica a")


 • Endocardite verrucosa")












 1. 2. 3. 4. 5. 6. 7. 8. Rash malare 9.")



 LES attivo, nefrite Infezioni Morte")
































 • Anticorpi anti-Ro/SSA • Anticorpi anti-La/SSB • Fattore reumatoide")
















 o ipopigmentate(falsa vitiligine)")

















 con valori di CPK non")










 Dolore e debolezza")












: dispnea, tosse e talora dolore toracico")










- Slides: 191
CONNETTIVITI –Lupus eritematoso sistemico –Sindrome da anticorpi anti-fosfolipidi –Sindrome di Sjögren –Sclerodermia –Polimiosite/dermatomiosite –Crioglobulinemia mista –Vasculiti sistemiche
Connettiviti • Malattie autoimmuni sistemiche • Spesso caratterizzate autoanticorpi dalla presenza di • Eziologia multifattoriale • Decorso caratterizzato remissioni da esacerbazioni e • Quadri clinici differenti, nell’ambito della stessa sindrome (sia in uno stesso paziente che tra pazienti) • Possibile sovrapposizione tra le varie sindromi
GM, femmina, 25 anni • Da due anni durante l’inverno le sue mani diventano fredde e le dita cambiano colore. • Riferisce che prima diventano bianche, poi viola, poi rosse. • Tale situazione si associa alla presenza di dolore. • Viene perché quest’anno il fenomeno si è accentuato e compare anche quando il freddo non è molto marcato. Forse ha avuto un paio di episodi in estate entrando in acqua al mare
Fenomeno di Raynaud
Il fenomeno di Raynaud (Maurice Raynaud 1898) • Ischemia acrale parossistica, scatenata dal freddo o dalle brusche riduzioni della temperatura, che si manifesta con una variazione del colorito cutaneo • In genere compare il pallore, seguito dalla cianosi e quindi dalla comparsa di un colorito rosso cupo, ma possono manifestarsi anche la sola fase di sbiancamento o la cianosi in assenza della fase di arrossamento
Epidemiologia • Colpisce tra il 5 e il 15% della popolazione generale • Particolarmente frequente nelle giovani donne
Quadro clinico • Il fenomeno di Raynaud coinvolge prevalentemente le estremità degli arti superiori ed inferiori • Nella maggior parte dei casi è bilaterale e simmetrico • In circa i due terzi dei pazienti si ritrovano le classiche fasi di colorazione: pallore, cianosi, rossore • La comparsa è di solito repentina, mentre la durata è variabile
Fenomeno di Raynaud
Fenomeno di Raynaud
Fenomeno di Raynaud
Fenomeno di Raynaud
Classificazione • Primario: quando compare in assenza di malattie ritenute capaci di causarlo • Secondario: quando rappresenta il sintomo di una sottostante malattia, per esempio una malattia autoimmune sistemica
Connettiviti con fenomeno di Raynaud secondario • Sclerodermia sistemica • Connettivite mista • Poli-dermatomiosite • Lupus eritematoso sistemico • Sindrome di Sjögren • Vasculiti
Il lupus eritematoso sistemico
FC, femmina, 30 anni • È sempre stata bene, non riferisce patologie significative. Due gravidanze portate a termine senza problemi. • Due mesi fa comparsa di febbre non elevata, intensa stanchezza, ingrossamento dei linfonodi del collo. • Il curante pensa ad un fatto infettivo intercorrente ma il quadro clinico persiste e la signora è sempre più stanca, la febbricola persiste come la linfoadenopatia. • Esegue degli accertamenti che evidenziano leucopenia, ipergammaglobulinemia, aumento della VES. Una ecografia addominale mostra una linfoadenopatia diffusa. • Il cutante teme che la signora abbia una malattia ematologica e la invia a visita specialistica ematologica.
Valutazione ematologica • L’esame obiettivo evidenzia la presenza di linfoadenopatia. • Esami ematochimici: – Leucopenia, anemia, ipergammaglobulinemia, ipocomplementemia – Positività anticorpi anti-nucleo, anti-SSA • Biopsia linfonodale: linfonodi reattivi
Valutazione reumatologica • L’ematologo sospetta che la signora abbia una malattia autoimmune sistemica e la invia dal reumatologo • A questo punto (2 mesi di malattia) la signora riferisce anche la presenza di dolore a mani e polsi e sono evidenti segni di ARTRITE • L’esame obiettivo mostra: linfoadenopatia, rash malare, artrite di mani e polsi • Ulteriori indagini evidenziano: positività del test di Coombs diretto ed indiretto
Riassumendo: • • • Donna giovane che presenta: Sintomi costituzionali: febbre e astenia marcata Linfoadenopatia Artrite di mani e polsi Rash malare Anemia emolitica, leucopenia, ipocomplementemia, ipergammaglobulinemia • Positività di autoanticorpi non organo specifici: anticorpi anti-nucleo, anticorpi anti-ENA • DIAGNOSI: LUPUS ERITEMATOSO SISTEMICO
Quanti tipi di “LUPUS”? • Lupus eritematoso CUTANEO: – Lupus discoide – Lupus subacuto • Lupus eritematoso SISTEMICO
Quanti tipi di “LUPUS”? • Lupus eritematoso CUTANEO: – Lupus discoide – Lupus subacuto • Lupus eritematoso SISTEMICO
Il lupus eritematoso sistemico • Malattia infiammatoria, sistemica, cronica ad eziologia multifattoriale e patogenesi autoimmune • Caratterizzato dalla produzione di anticorpi antinucleo rivolti verso diverse specificità antigeniche • Caratterizzato da un quadro clinico estremamente eterogeneo
Dati storici XVIII secolo Il termine “lupus” viene usato per descrivere lesioni cutanee 1872 Kaposi descrive la natura sistemica del lupus eritematoso 1948 Hargraves descrive il fenomeno LE
Epidemiologia M/F 1/10 Picco Età fertile Prevalenza 15 -67/100, 000 abitanti • Caucasici • 20. 3/100, 000 • Asiatici • 48. 8/100, 000 • Afro-Caraibici • 207/100, 000 Incidenza 1. 8 -7. 6/100, 000/anno
Cosa causa il LES? • EZIOLOGIA IGNOTA!!!! • Si ritiene che si tratti di una eziologia multifattoriale • Sono coinvolti fattori individuali di ordine genetico ed endocrino e fattori ambientali distinti in infettivi (virus? ), fisici (raggi UV), chimicofarmacologici
Fattori genetici • Importanza evidenziata da studi familiari – concordanza tra gemelli omozigoti fino al 50% (negli eterozigoti 2 -8%) – Il 10 -16% dei pazienti ha parenti affetti da LES o da altre malattie autoimmuni • Differente prevalenza razziale • Associazione con HLA di classe II. In molte popolazioni il LES è associato al DR 2 e al DR 3 • Associazione con HLA di classe III. La prevalenza del LES è particolarmente elevata in soggetti con deficit congeniti del complemento, tra i quali i più comuni sono quelli delle frazioni C 2, C 4
Fattori ormonali • Ormoni sessuali: netta prevalenza del LES nel sesso femminile in età fertile • Gli estrogeni sono immunostimolanti, mentre gli androgeni sono immunosoppressori. Questo spiegherebbe la maggiore suscettibilità delle donne alle malattie autoimmuni • Fattori neuroendocrini: alterazioni della funzione ipotalamica; le più importanti sembrano essere un aumento della produzione di prolattina e una riduzione della produzione di cortisolo in risposta allo stress
Fattori ambientali • Agenti infettivi: virus? Batteri? è possibile che possano indurre la malattia in soggetti predisposti o indurne una riacutizzazione • Raggi ultravioletti: l’esposizione ai raggi UV altera la struttura del DNA e aumentandone la immunogenicità. Nel LES è presente fotosensibilità e sono descritti casi di riacutizzazione o anche di esordio della malattia a seguito della esposizione ai raggi UV nei mesi estivi • Farmaci: LES da farmaci causato dalla assunzione di farmaci come la clorpomaziona, la idralazina, la isoniazide, l’alfametildopa, la procainamide e la penicillamina • Cosmetici? Additivi? Coloranti? Conservanti?
Quadro clinico • Colpisce prevalentemente le donne giovani • Estremamente polimorfo, differenti quadri TRA pazienti e nello stesso paziente nel corso del follow up • Alternanza di fasi di remissione e fasi di riacutizzazione
Modalità di esordio • Sintomi costituzionali: astenia, anoressia, dimagrimento, febbre • Manifestazioni cutanee: rash malare • Manifestazioni articolari: artrite simmetrica a carico di mani e polsi • Sierositi: pleurite e pericardite • Manifestazioni ematologiche: anemia emolitica, leucopenia, piastrinopenia
Manifestazioni cliniche del LES Pisa (n= 500) (%) Letteratura (%) Artralgie 82 53 -95 Artrite 69 53 -95 Alopecia 63 3 -45 Fotosensibilità 60 11 -45 Anemia 58 30 -78 Rash malare 55 39 -61 Impegno renale 53 31 -60 Fenomeno di Raynaud 49 18 -58 Leucopenia 42 41 -66 Vasculite cutanea 35 21 -37 Sierosite 29 31 -63 Febbre 26 41 -86 Piastrinopenia 23 7 -45 Linfoadenopatia 12 10 -59
Manifestazioni generali • Spesso sintomi di esordio della malattia • Astenia: presente in oltre l’ 80% dei casi • Anoressia e calo ponderale • Febbre, non infettiva
Manifestazioni cutanee • Si osservano nel 75 -90% dei pazienti affetti da LES • Sono distinte in manifestazioni lupus specifiche e lupus non specifiche • Le lesioni non lupus specifiche sono osservabili in altre malattie autoimmuni. La loro presenza è suggestiva di malattia sistemica
Manifestazioni lupus specifiche • Lesioni acute: rash malare, eritema generalizzato, lupus bolloso • Lesioni subacute: anulari policicliche, psoriasiformi • Lesioni croniche: lupus discoide, lupus profundus
Lesioni acute: Rash malare • Lesione più comunemente osservata • Associato alla malattia sistemica • Rash eritematoso localizzato nella regione Generalmente risparmia le pieghe nasolabiali • Regredisce senza lasciare esiti malare.
Lesioni acute: Rash generalizzato
Lupus subacuto • Varianti: papulosquamosa, psoriasiforme e anulare/policiclica • Associato a fotosensibilità • Le lesioni sono localizzate più frequentemente su braccia, spalle, scollo, dorso • Il volto è coinvolto molto raramente • Anticorpi anti-Ro/SSA sono presenti nel 50 -60% dei casi • Le lesioni guariscono tipicamente senza lasciare cicatrici, ma possono essere osservate ipopigmentazionne e teleangectasie • Il 50% circa dei pazienti soddisfa i criteri per la malattia sistemica
Lupus subacuto, variante anulare
Lupus subacuto, variante papulosquamosa
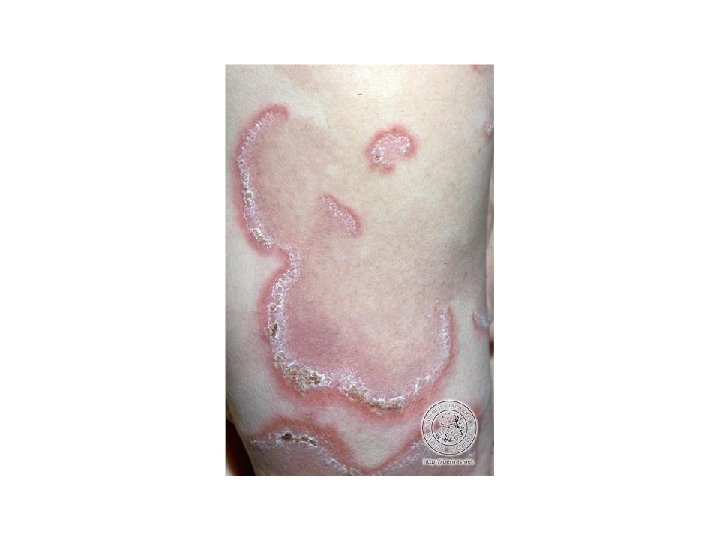
Lupus discoide • • Detto anche lupus cutaneo cronico Lesioni eritematose, rilevate, indurate, aderenti Localizzato: volto e collo, aree fotoesposte Generalizzato: più raro, caratterizzato dalla presenza di lesioni sopra e sotto il collo. Più spesso associato alla malattia sistemica • Possono svilupparsi atrofia cicatriziale, dispigmentazione, alopecia cicatriziale irreversibile • Nel 5% dei casi si osserva una evoluzione verso la malattia sistemica
Lupus discoide
Lupus discoide: impegno del cuoio capelluto con alopecia cicatriziale
Tipo di lesione Lupus acuto Lupus subacuto Lupus discoide Fine, facilmente staccata Fine, facilmente staccata Spesso, aderente Atrofia follicolare Assente Presente Fotodistribuzione Presente Marcata Presente Cicatrice Assente Presente Atrofia Assente Presente Lieve Marcata Presenti Aderenza Alterazioni pigmentarie Teleangectasie
Manifestazioni non lupus specifiche • Vascolari: vasculite, teleangectasie, livedo reticularis, ulcere croniche, gangrena periferica, noduli reumatoidi , • Lesioni orali, mucose • Alopecia non cicatriziale, frontale o diffusa • Panniculite • Lesioni orticarioidi • Sclerodattilia • Anomalie pigmentarie • Calcinosi cutanea
Manifestazioni non lupus specifiche Vasculite cutanea
Manifestazioni non lupus specifiche Teleangectasie
Manifestazioni non lupus specifiche Livedo reticularis: associata alla presenza di ab anti-fosfolipidi
Manifestazioni muscoloscheletriche • Artrite: tumefazione modesta a carico dei tessuti molli, non erosiva. Possono svilupparsi deformità (artrite di Jacoud( • Miosite: può essere secondaria alla flogosi articolare, alla terapia steroidea o dovuta ad una infiammazione delle fibre muscolari • Fibromialgia: sindrome dolorosa da stress secondaria a molte malattie reumatiche
Manifestazioni muscoloscheletriche • Impegno articolare: – Artrite – Artralgie – Coinvolgimento tendineo – Osteonecrosi • Miosite • Fibromialgia
Impegno articolare e LES • Artrite non erosiva • Artrite deformante non erosiva (artrite di Jaccoud) • Artrite erosiva (Rhupus) • Coinvolgimento tendineo (rottura tendinea dovuta alla infiammazione o all’uso di corticosteroidi) • Osteonecrosi
Artrite non erosiva • Si osserva nella maggior parte dei pazienti affetti da LES (fino al 90% nel corso della malattia) • Spesso manifestazione di esordio • Di solito artrite simmetrica, con distribuzione simile alla artrite reumatoide • Coinvolge principalmente: articolazioni delle mani (IFP), polso e ginocchia • La sinovite di solito è transitoria e reversibile
Artrite di Jaccoud • Artrite deformante che coinvolge prevalentemente le articolazioni metacarpofalangee, interfalangee prossimali ed i polsi • Sublussazioni articolari non erosive e riducibili • Assenza di erosioni ossee • Si osserva nel 10 -35% dei pazienti • Probabilmente legata alla presenza di sinovite cronica e al coinvolgimento di strutture periarticolari (capsula articolare, legamenti, tendini)
Jaccoud’s arthritis
Artrite erosiva- Rhupus • Rara, osservabile in meno del 5% dei pazienti • Rigidità mattutina (superiore a 1 ora) • Presenza di erosioni radiologicamente evidenziabili
Tendini • Degenrazione e rottura associate con: – terapia corticosteroidea, – prolungata durata di malattia – presenza di muscolocheletriche altre • Presentazione di solito acuta manifestazioni
Osteonecrosi • Necrosi ossea legata ad una alterato circolo apporto ematico • Incidenza variabile tra il 4 e il 18% • La testa femorale è coinvolta nell’ 80% dei casi • Dolore persistente, presente con il movimento, localizzato ad una singola articolazione • Possobili cause: – Corticosteroidi – Emboli grassosi, fenomeno di Raynaud, vasculite dei piccoli vasi, sindrome da anticorpi anti-fosfolipidi
Impegno renale nel LES • • • L'incidenza dell'impegno renale è stimata tra il 30 e l’ 80% nelle diverse casistiche Cambiamento della prognosi in termini di morbidità e mortalità Fino al 25% dei pazienti con impegno renale sviluppa insufficienza renale cronica • E’ caratterizzato dalla presenza di una infiammazione dei glomeruli (glomerulonefrite( • La classificazione della OMS distingue 6 diversi pattern di impegno renale nel LES
Modalità di esordio dell’impegno renale Sindrome nefrosica 45% Proteinuria persistente 48% Insuff renale acuta 5% Insuff renale cronica 2%
Classificazione WHO Quadro clinico associato Classe I: glomeruli normali Assenza di alterazioni Classe II: gn mesangiale Lieve proteinuria, modeste alterazioni del sedimento urinario Classe III: gn proliferativa focale Proteinuria raramente nefrosica, alterazioni del sedimento urinario Rara la alterazione della funzione renale Classe IV: proliferativa diffusa Proteinuria spesso nefrosica, ematuria, ipertensione, insufficienza renale Classe V: gn membranosa Sindrome nefrosica. Talvolta insufficienza renale a lenta evoluzione Classe VI: sclerosi glomerulare Insufficienza renale terminale
Glomerulonefrite proliferativa diffusa • Forma più comune, associata alla prognosi peggiore • La evoluzione clinica della glomerulonefrite proliferativa diffusa presenta una grande variabilità tra pazienti, nonostante apparenti somiglianze relativamente a variabili epidemiologiche, cliniche, sierologiche ed istologiche
Sierositi • Osservabili in oltre il 50% dei pazienti • Possono presentarsi come pleurite, pericardite, peritonite. • La pleurite in corso di lupus è bilaterale nel 50% dei casi. • I versamenti sono generalmente modesti. • La ascite si associa alla peritonite nell’ 11% dei casi circa; talvolta può divenire cronica.
Manifestazioni ematologiche • Anemia: anemia da malattia cronica (normocitica e normocromica), anemia emolitica a patogenesi autoimmune • Leucopenia: per lo più dovuta ad anticorpi antileucociti. • Linfopenia: attribuibili ad anticorpi linfocitotossici. • Piastrinopenia: attribuita ad anticorpi antipiastrine, ma anche elemento caratteristico della sindrome da anticorpi anti-fosfolipidi.
Impegno neuropsichiatrico • Prevalenza variabile tra il 20 e l’ 80% nelle differenti casistiche • Caratterizzato dalla presenza di manifestazioni neurologiche e/o psichiatriche • Difficoltà di inquadramento: – Estrema varietà delle manifestazioni cliniche – Mancanza di criteri classificativi uniformi – Incompletezza dei dati che derivano dalle tecniche di neuroimaging
Nomenclatura ACR: Sindromi neuropsichiatriche Sistema nervoso centrale Manifestazioni neurologiche • • Meningite asettica Malattie cerebrovascolari Sindrome demielinizzante Cefalea Disordini del movimento Mielopatia Convulsioni Myasthenia gravis Manifestazioni psichiatriche • • • Stato confusionale acuto Disturbi di ansia Disfunzione cognitiva Disturbi umore Psicosi Sistema nervoso periferico Poliradicolonevrite demielinizzante acuta Disordini autonomici Mono/polineuropatia Neuropatia nn cranici Plessopatia
Manifestazioni cardiache • Miocardite: da vasculite dei piccoli rami coronarici(? ) • Endocardite verrucosa atipica (Libman Sacks): endocardite verrucosa non batterica che raramente produce delle alterazioni emodinamicamente significative. • Coronaropatie: i pazienti con LES presentano una aterosclerosi accelerata in parte correlabile alla terapia farmacologica e in parte alla patologia stessa.
Miocardite • Manifestazione rara e spesso asintomatica, con una prevalenza variabile dall’ 8 al 25%. • Deve essere sospettata in caso di tachicardia a riposo, anomalie ECG, cardiomegalia. • L’ecocardiogramma può rivelare la presenza di anomali sistoliche e diastoliche del ventricolo sinistro. • La miocardite acuta si può associare ad altre manifestazioni di attività, in particolare alla pericardite
Pericardite, versamento pericardico • E’ la manifestazione cardiaca più frequente nel LES e raggiunge una prevalenza del 48% • Il versamento pericardico è di solito modesto • Normalmente la pericardite è asintomatica ed ha un decorso benigno • La complicanza più grave è una pericardite purulenta che potrebbe svilupparsi nel paziente immunosoppresso • Versamenti cospicui con tamponamento cardiaco e la pericardite restrittiva sono rari nel LES
Endocardite di Libman-Sacks Definizione: Presenza di vegetazioni verrucose aderente all’endocardio Patogenesi: Deposizione di immunoglobuline e complemento, con proliferazione e degenerazione cellulare e deposizione di fibrina; Correlazione con ACLA? Prognosi: Tipicamente è asintomatica Le vegetazioni possono frammentarsi a provocare embolizzazioni Generalmente buona, non valvulopatia Terapia: Nessuna
Manifestazioni polmonari del LES • Dolore toracico pleuritico • Polmonite lupica acuta • Polmonite interstiziale cronica • Sindrome “Shrinking Lung” • Ipertensione polmonare • Emorragia polmonare
Dolore toracico pleuritico Dolore muscoloscheletrico Spesso muscolare, o a carico delle articolazioni costo-condrali (sindrome di Tietze) Risponde al calore locale, analgesici topici, anti-infiammatori non steroidei Pleuritico Difficile da diagnosticare Possono essere presenti sfregamenti o versamento pleurico Il versamento di solito è modesto, bilaterale nel 50%
Polmonite lupica acuta • Rara, incidenza 1 -12% • Caratterizzata da febbre, tosse, emottisi, dispnea, pleurite, nel 50% dei casi si associa a versamento pleurico • Infiltrati polmonari generalmente basilari • Non è possibile isolare dei patogeni • La prognosi generalmente è scadente • Mortalità a breve termine del 50% • Residuano anomalie della funzionalità respiratoria con difetti di tipo restrittivo
Polmonite lupica acuta • Caratterizzata dalla presenza da infiltrati polmonari diffusi, mono- o bilaterali che interessano prevalentemente le basi polmonari • Talvolta infiltrati polmonari basilari possono essere secondari ad emorragie alveolari.
Polmonite cronica • Si osserva fino al 9% dei pazienti • Spesso preceduta da polmonite cronica • Più spesso in pazienti con malattia di lunga durata • Tosse cronica non produttiva, dispnea e dolore toracico pleuritico • Pattern restrittivo alle prove di funzionalità respiratoria con ridotti volumi polmonari
Shrinking lung • Sindrome restrittiva in assenza di alterazioni parenchimali, dovuto al sollevamento degli emidiaframmi (miopatia(? • Alla radiografia del torace possono essere presenti aree di atelectasia basilare
Manifestazioni gastrointestinali • • Vasculite mesenterica Malattie infiammatorie dell’intestino Morbo celiaco Epatite lupoide
Anticorpi antinucleo e LES Associazioni Sensitività Specificità 57 97 Anti-ds. DNA LES Anti-SSB-La Sjögren, subacute cutaneous lupus, neonatal lupus 16 -40 94 Anti-SSA-Ro Sjögren, subacute cutaneous lupus, neonatal lupus 8 -70 87 Anti-Sm LES 25 -30 elevata Anti-RNP MCTD 12 96
Come si fa diagnosi di LES? • Manifestazioni cliniche • Esami ematochimici • Presenza di autoanticorpi • CRITERI CLASSIFICATIVI
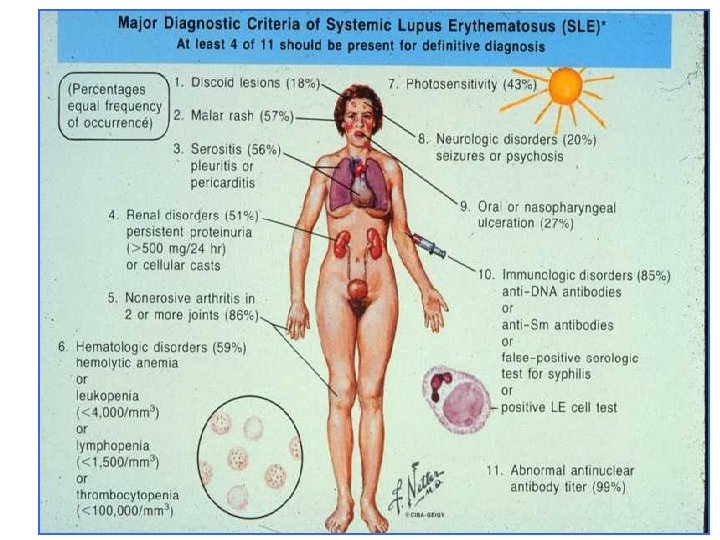
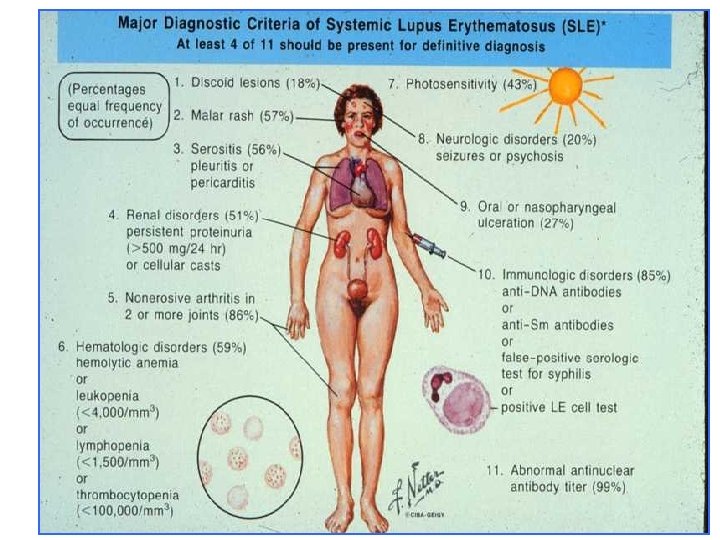
Criteri ACR (1982) 1. 2. 3. 4. 5. 6. 7. 8. Rash malare 9. Alterazioni ematologiche Rash discoide 10. Alterazioni Fotosensibilità immunologiche: cellule Aftosi orale LE, falsa positività STS, Artrite anti-ds. DNA, anti-Sm Pleurite/ pericardite 11. Anticorpi antinucleo Impegno renale Impegno neurologico
Prognosi • Netto miglioramento della sopravvivenza
Sopravvivenza a 5 e 10 anni • Ridotta mortalità • Maggiore attenzione su morbidità: ü danno ü comorbidità ü qualità della vita
Prognosi • Sviluppo di danno cronico: – 20 % circa dei pazienti con impegno renale sviluppa insufficienza renale – Lesioni neurologiche permanenti – Esiti cicatriziali di impegno cutaneo
Pattern di mortalità bimodale Morte precoce (entro 5 anni) LES attivo, nefrite Infezioni Morte tardiva LES inattivo Nessuna infezione IMA Urowitz et al Am J Med 1976
Cause di morte La attività di malattia come causa di morte nel 26% dei casi in associazione ad altri fattori (impegno di organo, infezioni) Nossent et al, Lupus 2007
Sindrome da anticorpi anti -fosfolipidi
AB, femmina, 34 anni • Giunge alla nostra osservazione perché ha avuto 4 aborti consecutivi tutti tra la 8 e la 10 settimana di gravidanza. • Il ginecologo ha consigliato valutazione reumatologica (tra le altre indagini…) • All’esame obiettivo è evidente livedo reticularis agli arti inferiori
Livedo reticularis: associata alla presenza di ab anti-fosfolipidi
Indagini • Ricerca di autoanticorpi non organospecifici: • Positività di anticorpi anti-cardiolipina a titolo elevato • Positività di anticorpi anti-B 2 GPI • DIAGNOSI: SINDROME DA ANTICORPI ANTI-FOSFOLIPIDI
BB, femmina, 19 anni • Da due giorni intensa cefalea • Oggi viene portata al PS per la comparsa di emiparesi sinistra ed alterazioni del sensorio • RM eseguita in urgenza mostra la presenza di trombosi della arteria cerebrale media • Esami ematochimici: piastrinopenia, positività LAC, anti-cardiolipina, anti-B 2 GPI • DIAGNOSI: SINDROME DA ANTICORPI ANITFOSFOLIPIDI
La arteria cerebrale media è il ramo principale che origina dalla carotide. La arteria rifornisce una parte del lobo frontale e la superficie laterale dei lobi temporale e parietale, incluse le aree motoria e sensitiva della faccia, gola, braccio e, nell’emisfero dominante, della parola La arteria cerebrale media è colpita molto frequentemente nell’ictus
Sindrome da anticorpi antifosfolipidi • Malattia caratterizzata da: – trombosi arteriose e/o venose ricorrenti – aborti ripetuti – Trombocitopenia • In associazione a titoli medio-alti di anticorpi antifosfolipidi.
Manifestazioni cliniche principali
Trombosi • Gli eventi trombotici sono spesso episodici e imprevedibili • Sono frequenti eventi ripetuti • Trombosi venose e/o arteriose, possono realizzarsi in qualsiasi distretto • Le trombosi venose sono più frequenti di quelle arteriose • Spesso una trombosi arteriosa tende ad essere seguita da una nuova trombosi arteriosa e una trombosi venosa da una nuova trombosi venosa • I fattori che predispongono verso una trombosi arteriosa o una venosa non sono noti
Trombosi venosa • Potenzialmente interessato qualsiasi vaso venoso • Le vene superficiali e profonde degli arti inferiori sono interessate più frequentemente • Altre sedi di trombosi venosa: vene renali, mesenteriche, porta, epatica, retiniche, vena cava inferiore
Trombosi arteriosa • La sede più frequente di trombosi arteriosa sono i vasi cerebrali • La occlusione di arterie cerebrali si associa a manifestazioni neurologiche gravi quali ictus, TIA, epilessia, cefalea intrattabile • Altre sedi di trombosi arteriosa: arterie retiniche, mesenteriche, coronarie, renali
Manifestazioni neurologiche • Le manifestazioni neurologiche in corso di SAPL sono molteplici. Alcune quali l’ictus, l’epilessia, la demenza, la mielite trasversa, e la amaurosi fugace sono correlate direttamente allo sviluppo di trombosi o ad embolizzazioni • Altre manifestazioni neurologiche, come la amnesia transitoria, la depressione, disordini del movimento (corea) non hanno una chiara correlazione patogenetica con gli anticorpi anti-fosfolipidi
Manifestazioni neurologiche • Ischemia focale • Disordini del movimento – attacchi ischemici transitori – infarti cerebrali • Occlusione multipla piccole arterie di – demenza multi-infartuale – encefalopatia ischemica acuta • Trombosi venosa cerebrale • Ictus embolico – corea – atassia cerebellare • Sindromi spinali – mielite trasversa – sindrome di Guillain Barrè • Cefalea • Epilessia
Gravidanza • Poliabortività • Perdita fetale • Ritardo di accrescimento • Parti pretermine • Si osservano spesso trombosi dei vasi placentari ed infarti placentari • Aumentata incidenza di complicanze materne: gestosi, eclampsia
Altri manifestazioni della sindrome da anticorpi anti-fosfolipidi • Trombocitopenia • Ulcere agli arti inferiori • Livedo reticularis • Anemia emolitica • Manifestazioni neurologiche quali ischemia cerebrale transitoria, mielite trasversa, corea, emicrania, deficit di memoria
Livaedo
Prognosi • Difficile da valutare e correlata al tipo di impegno di organo • L’uso degli anticoagulanti “a vita” hanno drasticamente ridotto la incidenza di trombosi ripetute • La prognosi è cattiva in quei rari casi di pazienti con episodi trombotici ricorrenti nonostante la terapia anticoagulante
Sindrome di Sjögren
PP, femmina, 45 anni • Viene inviata dal dentista che ha notato che la signora sviluppa carie con eccessiva frequenza, nonostante una adeguata igiene orale e cure. • Anamnesi: la signora riferisce di avere smesso di usare le lenti a contatto per intolleranza (secchezza degli occhi). Riferisce anche secchezza della cute.
Sindrome di Sjögren • Malattia infiammatoria, sistemica, cronica ad eziologia multifattoriale e patogenesi autoimmune • Decorso lentamente progressivo • Interessa principalmente le ghiandole esocrine, in particolare ghiandole salivari e lacrimali • Colpisce principalmente il sesso femminile (M: F=1: 9), picco di incidenza 40 -50 anni • Prevalenza: 6 -27/100, 000 abitanti
Epidemiologia
Manifestazioni cliniche • Secchezza delle mucose : xerostomia, xeroftalmia • Tumefazione delle ghiandole salivari maggiori • Poliartrite non erosiva • Fenomeno di Raynaud • Anomalie sierologiche: aumento della VES, anemia, leucopenia, linfopenia, iperamilasemia • Manifestazioni extraghiandolari: polmoni, reni, fegato • Aumentato rischio di linfomi
Manifestazioni di esordio
SINDROME DI SJÖGREN = SECCHEZZA
Impegno oculare • Talvolta subclinico, si manifesta con irritazione attribuita a congiuntivite batterica. • La manifestazione più tipica è la cheratocongiuntivite secca che comporta una distruzione dell’epitelio congiuntivale. • Le forme non trattate possono comportare gravi complicazioni che comprendono congiuntivite secondaria, ulcerazione corneale e perforazione. • La perforazione corneale può, a sua volta, causare uveite, cataratta e glaucoma.
Cheratocongiuntivite
Impegno orofaringeo • Xerostomia con difficoltà alla deglutizione di cibi secchi, cambiamento della percezione dei sapori, bruciore nel cavo orale, difficoltà a parlare a lungo, incremento delle carie dentali, candidiasi secondaria. • Tumefazione parotidea e delle ghiandole salivari maggiori: presente nel 60% dei casi, nella maggior parte dei pazienti è episodica, ma può anche divenatre persistente. La tumefazione inizialmente può essere unilaterale ma più spesso è bilaterale.
Sindrome di Sjögren: tumefazione parotidea
Manifestazioni extraghiandolari
Malattie linfoproliferative • Rischio relativo di sviluppare un linfoma di 44 volte superiore rispetto alla popolazione generale • Principalmente linfomi originanti dai linfociti B • La proliferazione dei linfociti B si può presentare come macroglobulinemia di Waldenstrom o come gammopatie monoclonali non Ig. M
Anomalie sierologiche • Aumento della VES • Anemia • Leucopenia, linfopenia • Ipergammaglobulinemia policlonale • Iperamilasemia
Autoanticorpi • Anticorpi antinucleo (ANA) • Anticorpi anti-Ro/SSA • Anticorpi anti-La/SSB • Fattore reumatoide 95 -100% 50 -90%
Conclusioni • La sindrome di Sjögren è una malattia “benigna”. • I sintomi principali secchezza. sono correlabili alla • L’impegno di organi vitali è raro. • Raramente i pazienti con sindrome di Sjögren assumono terapie aggressive.
SCLERODERMIA
CS, 48 anni • Da due anni le sue mani diventano bianche con il freddo, sono estremamente dolenti e gonfie • Da due mesi ha alcune ulcere dolenti sui polpastrelli delle dita delle mani • Ha notato un progressivo indurimento della pelle delle mani • Inoltre ha la sensazione che la sua bocca si apra con difficoltà • Da un anno ha disfagia ed è dimagrita di 4 Kg
Definizione • Il termine sclerodermia identifica alcune malattie caratterizzate dalla fibrosi (un abnorme aumento del tessuto connettivo) nella cute e in altri apparati • La fibrosi è conseguente ad alterazioni funzionali ed organiche del microcircolo e a complesse anomalie della funzione immunitaria
Epidemiologia • Prevalenza: 1500 casi/1, 000 • Incidenza: 10 -20 casi/1, 000 • Non riportate differenze razziali • M: F= 1: 3 fino a 1: 7 • Picco di incidenza 30 -50 anni
Classificazione basata sulla estensione dell’impegno cutaneo Sclerodermia diffusa Interessamento cutaneo esteso al tronco oltre che arti e volto Sclerodermia limitata Sclerosi cutanea limitata a mani, piedi, volto, avambracci Sinonimo CREST: Calcinosi, Raynaud, Esofago, Sclerodattilia, Teleangectasie Sclerodermia sine scleroderma Senza impegno cutaneo, ma con impegno degli organi interni
Manifestazioni cliniche in 1012 pazienti italiani Raynaud 96% Teleangectasie 69% Impegno esofageo 60% Impegno polmonare 60% Ulcere cutanee 48% Sindrome secca 33% Impegno cardiaco 30% Ipermelanosi 21% Calcinosi 21% Artrite 19% Impegno renale 7%
Quadro clinico • Fenomeno di Raynaud • Indurimento ed inspessimento della cute • Coinvolgimento di organi interni, incluso il tratto gastrointestinale, i polmoni, il cuore, i reni • Il rischio di coinvolgimento degli organi interni è fortemente correlato alla estensione e progressione dell’impegno cutaneo
Esordio • La sclerosi sistemica esordisce nella maggior parte dei casi con il fenomeno di Raynaud • Il fenomeno di Raynaud si manifesta pochi mesi prima della sclerosi cutanea nella forma diffusa, può precedere di mesi o anni le manifestazioni cutanee nella forma limitata • Raramente la malattia può esordire con manifestazioni articolari, con una miosite o con una manifestazione viscerale (dispnea, disfagia)
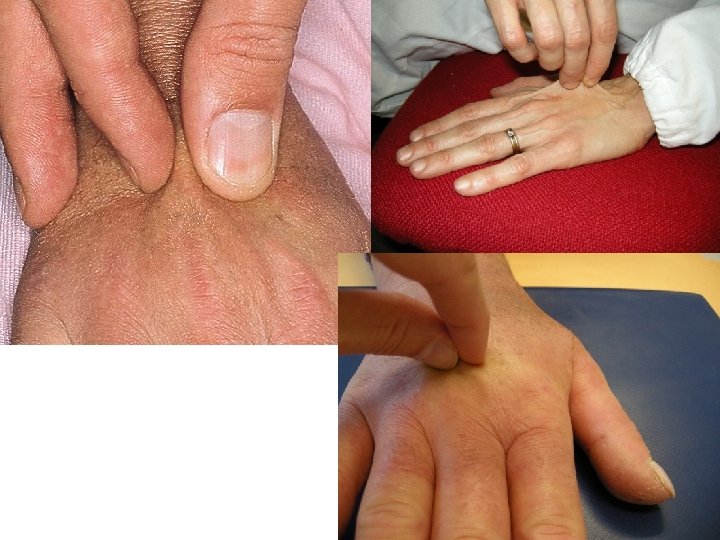
Sclerosi cutanea • Evolve in tre fasi: – Fase edematosa: la cute appare ispessita, le dita rigide e tumefatte – Fase sclerotica: la cute è di consistenza aumentata, poco elastica e non sollevabile in pliche – Fase atrofica: la cute appare assottigliata • Il processo fibroso colpisce anche le ghiandole sudoripare e i bulbi piliferi con caduta di peli e capelli • A carico del volto si sviluppa la tipica facies sclerodermica, amimica
Sclerosi cutanea • Nella forma diffusa può essere coinvolta qualsiasi area del corpo • Nella forma limitata sono interessati solo mani, avambracci, piedi e volto • L’impegno cutaneo è attivo nei primi 5 anni di malattia • Dopo i 5 anni si stabilizza e la cute può ammorbidirsi leggermente
Sclerosi cutanea con difficoltà a sollevare la cute in pliche
Sclerosi cutanea diffusa
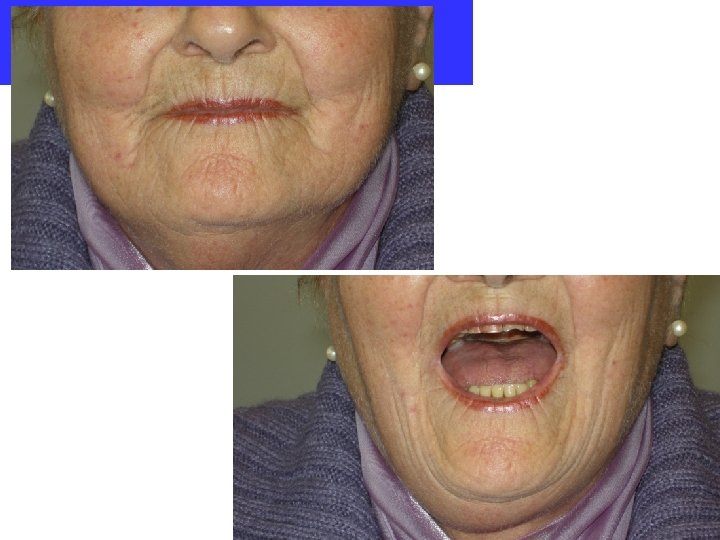
Facies sclerodermica Elementi sono: caratteristici • le labbra sottili • la rima orale che si apre poco e non si chiude completamente • il naso affilato • le rughe della fronte spianate
Facies sclerodermica
Sclerodermia, particolare della rima buccale
Altre manifestazioni cutanee • Discromie: chiazze iperpigmentate talora molto estese (melanodermia) o ipopigmentate(falsa vitiligine) • Calcinosi: depositi grossolani di fosfato e carbonato di calcio, che spesso affiorano alla superficie cutanea e possono anche ulcerarsi • Prurito: causato da irritazione della cute attribuibile al processo infiammatorio sottostante, non associato ad eruzioni cutanee
Calcinosi delle dita delle mani
CREST-syndrome: calcificazioni sottocutanee.
Alterazioni vascolari • Fenomeno di Raynaud: 95% dei pazienti. Il vasospasmo è ben evidente a carico di mani e piedi ma può realizzarsi anche in altre sedi (reni, cuore, polmoni). • Con il tempo si sviluppano sulla cute ulcere e necrosi (20% dei pazienti). La perdita di tessuto può coinvolgere anche l’osso sottostante, con amputazione delle falangi. • Teleangectasie: dilatazioni di arteriole, capillari e venule che formano chiazze rosse di varie dimensioni. Si formano più frequentemente a carico di volto, collo, torace e mani.
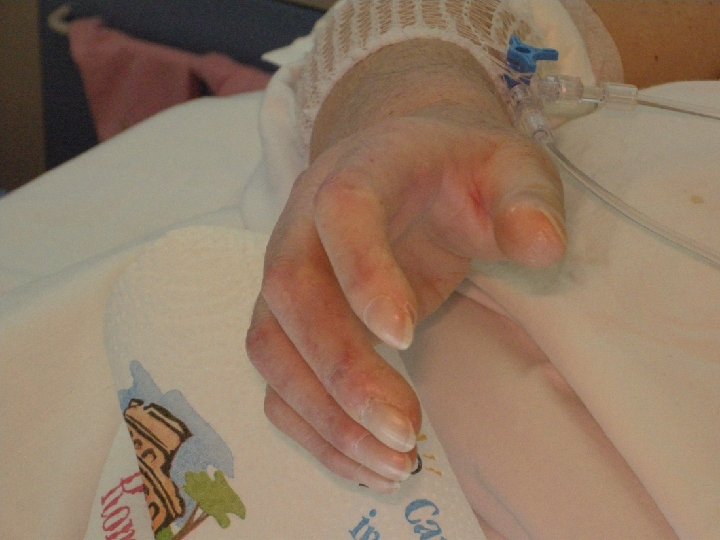
Ulcere cutanee • Sono causate dalle alterazioni circolatorie o da microtraumi • Generalmente si sviluppano sui polpastrelli o su prominenze ossee • Molto dolorose • Interferiscono con la funzione delle mani • Possono infettarsi
Sclerodermia: ulcere digitali
Sclerodermia: ulcere digitali
Sclerodermia: ulcere digitali
Sclerodermia: teleangectasie
Manifestazioni articolari • Artralgie/Artrite non erosiva, mono o oligoartrite • Tendiniti che provocano dolore e scrosci alla palpazione • Lesioni ossee: i fenomeni ischemici provocano osteolisi con riassorbimento osseo • Quando la sclerosi cutanea è estesa le mani vanno incontro ad una retrazione con la comparsa di deformità irreversibili (mani ad artiglio) • Contratture in flessione
Sclerodermia: sclerodattilia
Sclerodermia: sclerodattilia
Apparato gastrointestinale • Esofago: interessato nel 60 -75% dei casi. – Ipotonia ingravescente, con marcata alterazione e riduzione della peristalsi. – Reflusso gastro-esofageo ed esofagite – Disfagia, epigastralgie, pirosi • Stomaco e intestino: ridotta motilità, che porta a stipsi/diarrea, malassorbimento
Sclerodermia: ipotonia esofagea
Impegno polmonare • I polmoni sono colpiti nel 70 -80% dei pazienti • Forma diffusa: fibrosi interstiziale che comporta una progressiva compromissione della funzione respiratoria • Forma limitata: ipertensione polmonare determinata da una vasculopatia obliterante • Pleurite
Fibrosi polmonare
Fibrosi polmonare grave con ispessimento dei setti interlobulari
Impegno cardiaco • Pericardite: nel 33% dei pazienti si osserva un versamento pericardico che può essere acuto o cronico • Miocardiopatia restrittiva: possibile sviluppo di fibrosi in pazienti con spasmo coronarico (Raynaud coronarico) • Frequenti disturbi cardiaca e aritmie della conduzione
Impegno muscolare • Comune una modesta miosite (infiammazione muscolare) con valori di CPK non elevati • Miosite grave: si osserva in particolare nei casi di sindrome da “sovrapposizione” , i valori di CPK sono elevati
Impegno renale • Nella maggior parte dei casi lievi anomalie del sedimento urinario • Il quadro più severo è noto come crisi renale ed è caratterizzato da insufficienza renale rapidamente progressiva spesso associata a ipertensione arteriosa maligna • L’impegno renale è correlato alla proliferazione vascolare tipica della malattia
Anomalie sierologiche • Indici di flogosi: di solito sono scarsamente aumentati, per lo più in presenza di necrosi o infezioni di ulcere cutanee • Anemia: di tipo microcitico da malattia cronica oppure macrocitica da laterato assorbimento intestinale di vitamina B 12 e folati • Altre alterazioni bioumorali sono secondarie a impegni viscerali: – Enzimi muscolari: aumentano in caso di miosite – Azoto e creatinina: impegno renale
Autoanticorpi • Anticorpi anti-nucleo: positivi in oltre il 95% dei caso • Anticorpi specifici della SSC: – Anticorpi anti-centromero: caratteristici della forma limitata, nella quale si osservano nel 60 -70% dei casi – Anticorpi anti-topoisomerasi (Scl 70): caratteristici della forma diffusa, nella quale sono positivi nel 40% dei casi e si associano con alcuni impegni viscerali (cuore, polmone)
Decorso della forma diffusa • • • Esordio del fenomeno di Raynaud entro 1 anno dalla comparsa di sclerosi cutanea (o impegno cutaneo) Sclerosi cutanea diffusa a tronco ed estremità Comparsa precoce di interstiziopatia polmonare, insufficienza renale, impegno gastrointestinale diffuso, impegno miocardico Presenza di anticorpi anti-topoisomerasi I (anti-Scl 70) Prognosi peggiore con un decorso clinico più aggressivo e una sopravvivenza a 10 anni del 55% circa
Decorso della forma limitata • • Fenomeno di Raynaud che precede di anni l’esordio • Elevata incidenza tardiva di polmonare, calcinosi, teleangectasie • • Sclerosi cutanea limitata a mani, piedi, volto, avambracci Presenza di anticorpi anti-centromero Sopravvivenza del 70% a 10 anni ipertensione
Terapia • Farmaci attivi sul circolo: – calcio antagonisti – prostacicline • Immunosoppressori: – ciclosporina – ciclofosfamide – micofenolato • Antifibrotici: – penicillamina – colchicina
Miopatie idiopatiche infiammatorie
Miopatie infiammatorie • Gruppo eterogeneo di malattie muscolari acquisite caratterizzate da un processo infiammatorio a carico della muscolature scheletrica • Incidenza: 1 -12 casi/anno/milione • Prevalenza: 4 casi/100, 000
Classificazione • Polimiosite idiopatica • Dermatomiosite amiopatica • Dermatomiosite giovanile • Miosite associata a neoplasie • Miosite associata a connettiviti • Miosite a corpi inclusi
Epidemiologia Polimiosite dell’adulto Dermatomiosite dell’adulto Giovanile Associate a malattie autoimmuni sistemiche Associate a neoplasie Totale Proporzione dei pazienti 50% 20% 10% 10% Età alla diagnosi 45 40 10 35 60 45 Rapporto M: F 2: 1 1: 1 10: 1 1: 1 3: 1 Incidenza razziale (N: B) 5: 1 3: 1 1: 1 3: 1 2: 1 3: 1 Incidenza: 1 -12 casi/anno/milione Prevalenza: 4 casi/100, 000
Modalità di esordio Debolezza prossimale senza dolore (oltre 3 -6 mesi) Dolore e debolezza prossimali acuti o subacuti (oltre 3 -6 mesi) Debolezza prossimale e distale insidiosa (oltre 1 -10 anni) Solo mialgie prossimali 55% Solo rash dermatomiositico <1% 30% 10% 5%
Manifestazioni cliniche nel corso della malattia Manifestazioni muscolari Debolezza muscolare simmetrica e diffusa Contratture muscolari Manifestazioni cutanee Papule di Gottron Segno di Gottron Rash eliotropo Calcificazioni sottocutanee Manifestazioni cardiache Aritmie Scompenso congestizio Ipertrofia ventricolare Pericardite Manifestazioni polmonari Debolezza dei muscoli respiratori Fibrosi polmonare interstiziale (5 -10%( Manifestazioni articolari Manifestazioni gastrointestinali Lieve artrite non erosiva Difficoltà a deglutire Disfagia esofagea Reflusso
Impegno muscolare • Debolezza muscolare ingravescente, generalmente simmetrica a carico della muscolatura prossimale degli arti e dei cingoli • Esordio subdolo e la progressione muscolare avviene lentamente dell’impegno • Oltre ai cingoli possono essere interessati i mm del collo, respiratori, della deglutizione, della fonazione • I segni clinici più rilevanti sono: la riduzione della forza muscolare, la dolorabilità alla palpazione dei muscoli, le contratture e la atrofia muscolare
Distribuzione della debolezza muscolare • PM e DM causano la debolezza della muscolatura prossimale • I muscoli interessati più spesso sono quelli dei cingoli, degli arti superiori e del collo
…conseguenze dell’impegno muscolare……. • Difficoltà a: – – – Salire le scale Alzarsi da una sedia o dal letto Camminare Deglutire Accavallare le gambe Sollevare il capo dal cuscino • Voce nasale • Disfagia nel 10 -15% dei casi
Impegno cutaneo • Rash eliotropo: nel 25% dei pazienti con DM. Colorazione violacea delle palpebre superiori accompagnata talora a edema • Papule di Gottron: nel 30% dei pazienti con DM. Sono papule o placche eritematose o violacee, lievemente sopraelevate, presenti al di sopra delle sporgenze ossee (più spesso articolazioni metacarpofalangee, interfalangee prossimali e distali) • Calcificazioni sottocutanee: più frequenti nell’infanzia. Possono formarsi sulle fasce che ricoprono le masse muscolari e possono ulcerare la cute
Dermatomiosite: rash eliotropo
Dermatomiosite: rash eliotropo
Dermatomiosite Rash eliotropo ed edema del volto
Dermatomiosite: papule di Gottron
Dermatomiosite: papule di Gottron
Dermatomiosite: papule di Gottron
Impegno cardiaco • Anomalie elettrocardiografiche: disturbi della conduzione AV nel 50% dei casi • L’impegno del miocardio (miocardite) è raro, nei casi severi si può associare una insufficienza cardiaca congestizia • Pericardite
Impegno polmonare • Fibrosi polmonare interstiziale (5 -10%): dispnea, tosse e talora dolore toracico • Debolezza dei muscoli respiratori che può causare una insufficienza respiratoria • Polmonite ab ingestis determinata dalla incoordinazione dei muscoli della deglutizione
Alterazioni bioumorali • Aumento degli indici aspecifici di flogosi • Aumento degli indici di necrosi muscolare: il più rappresentativo è la creatin-fosfochinasi (CPK) • Anticorpi antinucleo
Prognosi • Morte del 50% dei pazienti non curati • Sopravvivenza del 90% a 5 anni nei casi curati, ma del 55% nei casi associati a neoplasie
Invalidità • Ogni riattivazione della malattia determina una perdita di forza muscolare e difficilmente il paziente ritorna ad avere il precedente livello di forza muscolare • Alcuni studi hanno mostrato che 1/3 dei pazienti affetti da miopatie ha un livello variabile di invalidità • La invalidità aumenta in relazione alla durata della malattia prima del trattamento
Terapia • Corticosteroidi ad elevato dosaggio • Immunosoppressori: utilizzati principalmente con la funzione di risparmiatori di steroidi – ciclosporina – azatioprina – methotrexate – ciclofosfamide • Immunoglobuline e. v. • Fisioterapia: da iniziare dopo la remissione
AC, maschio, 23 anni • Da alcuni mesi è stanco e ha debolezza muscolare ingravescente, in particolare a carico dei muscoli dei cingoli • Ha attribuito la sintomatologia allo stress per lo studio • Nelle ultime due settimane la debolezza è peggiorata e non riesce più ad alzarsi dalla sedia ed ha difficoltà a mangiare autonomamente, nella deglutizione e nel tenere sollevata la testa • Il curante prescrive esami del sangue “generali” • Risultati: aumento delle transaminasi (SGOT 280, SGPT 300), aumento del CPK (3000)
Vasculiti sistemiche
Le vasculiti sistemiche • Gruppo eterogeneo di sindromi cliniche caratterizzate dalla infiammazione delle pareti dei vasi sanguigni • Le vasculiti possono interessare qualsiasi vaso sanguigno considerevole sovrapposizione nei processi patologici • Le manifestazioni cliniche delle vasculiti sono essenzialmente il risultato della ischemia dei tessuti irrorati dai vasi sanguigni infiammati e di sintomi generali legati alla presenza di un processo infiammatorio sistemico
Classificazione delle vasculiti Tipo di vasculite Arterite di. Takayasu Arterite temporale Panarterite nodosa Sindrome di Churg Strauss Vasculite primitiva del SNC Granulomatosi di Wegener Poliangite microcscopica Sindrome di Behcet Sindrome di Kawasaki Granulomatosi linfomatoide Associate a connettiviti Vasculiti leucocitoclastiche Aorta e suoi rami AA grosso e medio calibro AA piccolo calibro Venule, arteriole
Classificazione delle vasculiti Capillari AA. di grosso e medio calibro Vene Piccole arterie Angite cutanea leucocitoclastica Porpora di Schonlein Henoch e crioglobulinemia Poliarterite microscopica Granulomatosi di Wegener e S. di Schurg Strauss Panarterite nodosa e Kawasaki Arterite temporale arterite di Takayasu
Sinossi principali vasculiti