CONGENTAL ADRENAL HYPERPLAZA YRD DO DR GLAY ILER


 is one of the causes of adrenal insufficiency in infancy")


, the clitoris")












- Slides: 26
CONGENİTAL ADRENAL HYPERPLAZİA YRD. DOÇ. DR. GÜLAY ÇILER ERDAG
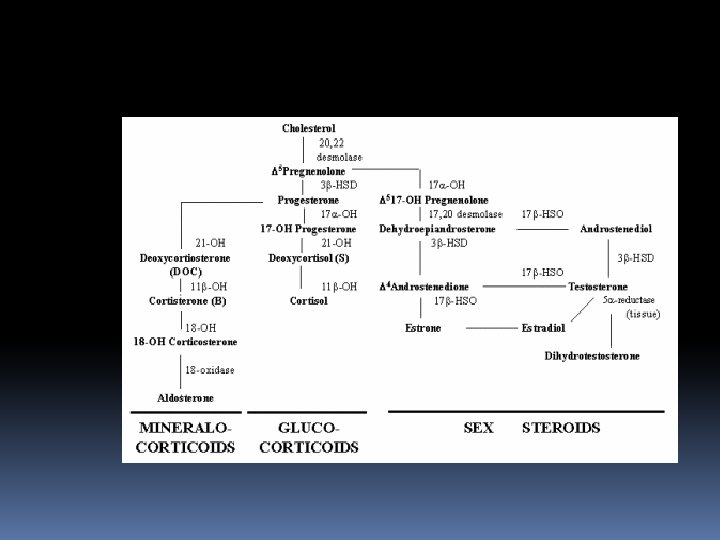
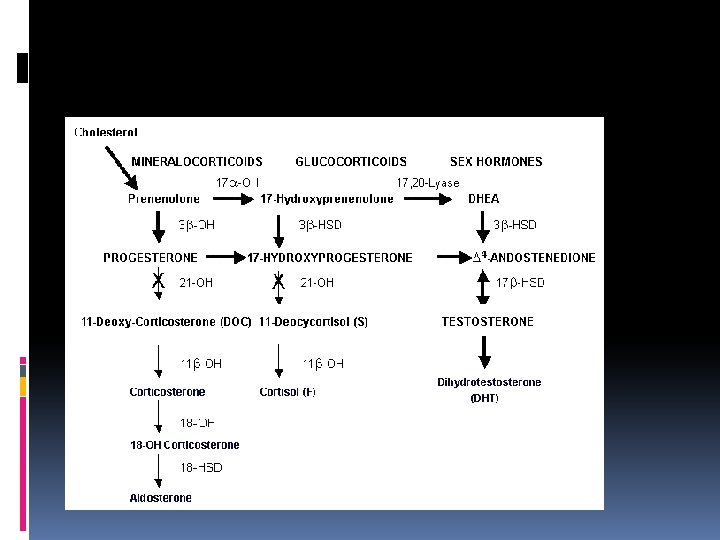
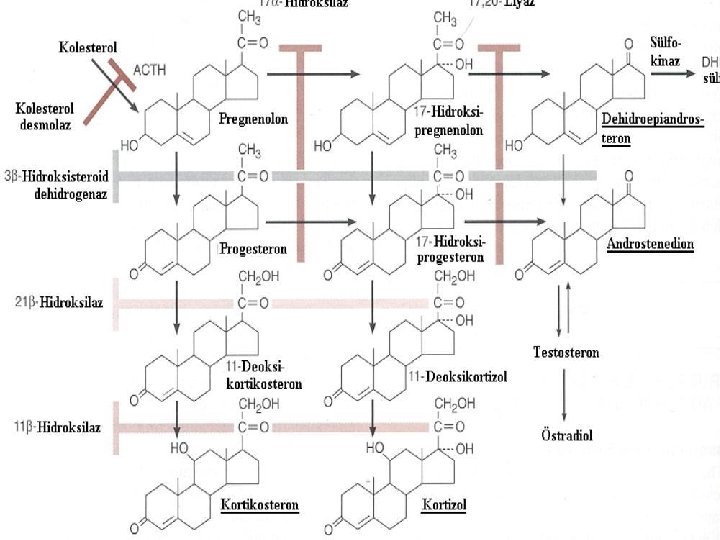
T The adrenal gland consists of: an outer cortex responsible for the synthesis of steroids an inner medulla synthesis catecholamines T T The adrenal cortex consist of three zones: v an outer GLOMERULOSA end product is the mineralocorticoid ALDOSTERONE(regulates sodium balance) v a middle zone FASCICULATA end product is CORTİSOL v an inner zone RETICULARIS synthesis SEX STEROIDS
Congenital adrenal hyperplasia(CAH) is one of the causes of adrenal insufficiency in infancy and childhood CAH is a disorder present at birth characterized by a deficiency in the hormones CORTISOL, ALDOSTERON and the overproduction of ANDROGEN
The different types of AGS are inherited as autosomal recessive gene defects This defect results in the lack of an enzyme needed by the adrenal gland to make cortisol In response to deficient cortisol the pituitary gland secretes ACTH that stimulates the adrenal gland: HYPERPLASIA
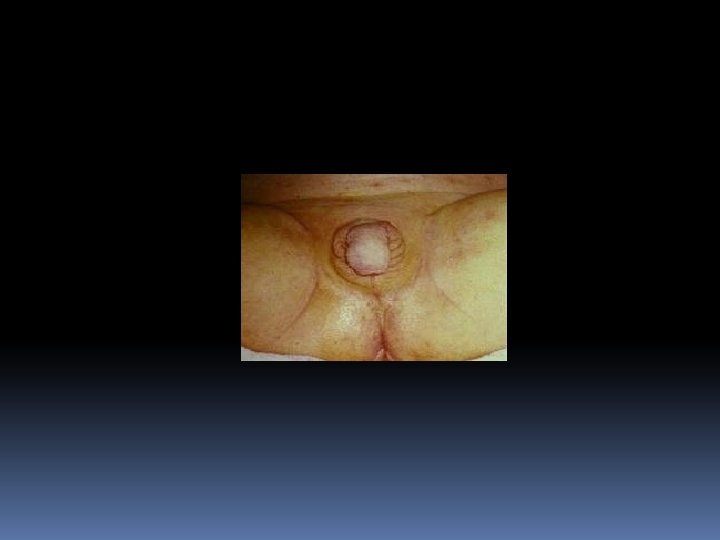
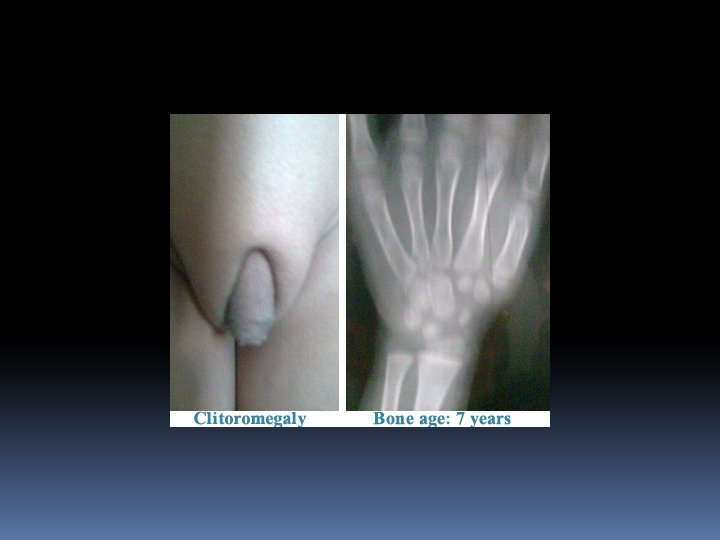
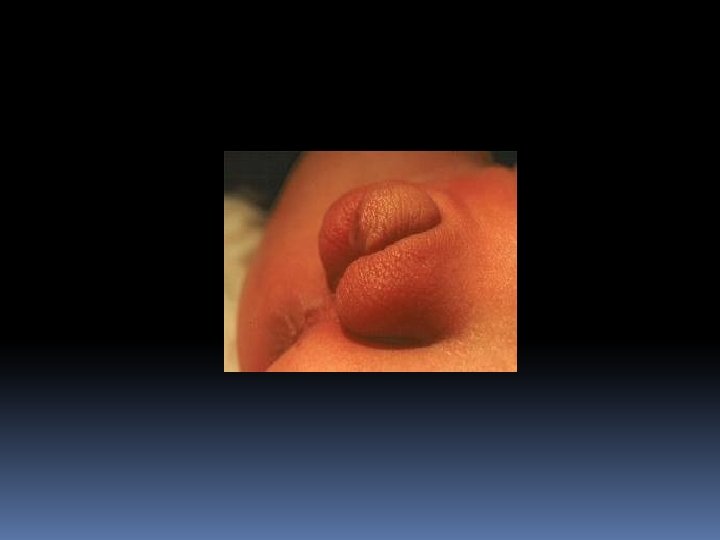
The condition affects both females and males In FEMALE newborn (pseudohermaphroditism), the clitoris is enlarged with the urethral opening at the base: ambigious genitalia-often appearing more male like than female The internal structures of the reproductive tract (ovaries, uterus and Fallopian tubes) are normal As she grows older masculinization of some features takes place: deepening of the voice, pubic hair before 2 nd birthday , appereance of facial hair, failur to menstruate at puberty
In a MALE newborn: The infant may appear normal at birth During the first few months of life the penis enlarges, the scrotum darkens, pubic hair appears and the voice deepens Affected males may appear to enter puberty as early as 2 -3 years of age. At puberty the testes are small and soft Bone age is advanced; Mental development is usually normal In BOTH- Height as chidren will be taller but ultimate adult height will be significantly shorter
Some forms of CAH are more severe and cause adrenal crisis in the newborn due to SALT WASTING In the salt losing form of AGS newborns develop symptoms shortly after birth (in the 1 st and 2 nd week): Vomiting Dehydration Electrolyte changes : Hyponatremia, hyperkalemia, metabolic acidosis Cardiac arrythmias Untreated, this condition can lead to death within 9 -14 days after birth Various types are recognized
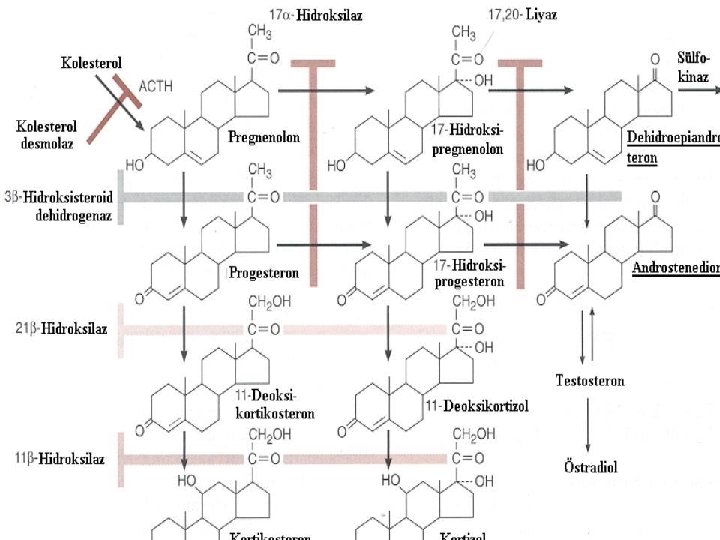
DEFICIENCY of 21 -HYDROXYLASE This enzyme, encoded by CYP 21 gene; Defects in CYP 21 cause congenital adrenal hyperplasia Inability to convert 17 -hydoxyprogesterone into 11 -deoxycortisol and of progesterone to desoxycorticosterone(DOC) Overproduction of testosterone makes the symptoms Biochemical diagnostic studies: Decreased urinary excretion of products of cortisol (17 hydroxycorticosteroids ) Elevated urinary 17 ketosteroids (excretion products of androgen pathway) Elevated serum testosterone whereas serum cortisol and aldosterone low Elevated 17 -hydoxyprogesterone-by the 3 rd day Elevated urinary pregnanetriol-major urinary metabolite of 17 OHP
There may be variable allelic forms of this disorder and other individual factors that result in variable expression of the defect both in terms of age at presentation This genetic and clinical heterogenecity has given rise to terms as: CLASSIC-salt wasting, simple virilizing form NONCLASSIC-affected person has a normal phenotype at birth; develop evidence of virilization during later childhood, adolescence or early adulthood ASYMPTOMATIC-no phenotypic features
DEFICIENCY of 11 -beta. HYDROXYLASE 5 -8% of cases Failure to convert 11 deoxycortisol to cortisol Associated with virilization and usually with hypertension (absent in the first few days of life) In blood 11 deoxycortisol levels are increased In urine 11 deoxycortisol, 17 KS are increased Classic-severe Nonclassic-milder
DEFICIENCY of 17 HYDROXYLASE Males-phenotypic females Females-failure of sexual development at the expected time Hypertension- overproduction of DOC Virilization, amenorrhea In blood: serum progesteron levels are increased In urine 17 KS, 17 OH corticoid are decreased
PARTIAL or COMPLETE DEFECT in 3 BETA HYDROXYSTEROID DEHYDROGENASE ACTIVITY 5% and less Failure to convert pregnenolone to progesteron In male: incomplete masculinization Hypospadias cryptorchidism In female: some degree of masculinization Severe sodium loss occurs Infant mortality rate is high in complete form In blood: Pregnenalone levels are increased In urine -17 KS, 17 OH corticoid levels are decreased
CHOLESTEROL DESMOLASE DEFICIENCY Clinical features are similar to those of 3 betahydroxysteroid dehydrogenase deficiency All steroid excretion is markedly decreased
TREATMENT The goal of treatment is to return the androgen hormone levels to normal This is achieved by daily administration of dexamethasone, fludrocortisone or hydrocortisone The gender of a baby with ambigious genitalia is determined by examination of the chromosomes
TREATMENT Reconstrictive surgery for girls with masculine external genitalia is usually performed between the ages of 1 -3 Medication to treat this disorder must be continued for life
Prenatal diagnosis is available for some forms of AGS It is accomplished in the first trimester: by chorionic villus sampling in the second trimester: by measuring hormones such as 17 OHP in the amniotic fluid
A newborn screening test is available for the most common form of AGS which can be done on heel stick blood (microfilter paper tecnique to measure 17 0 HP Rapid chromosomal diagnosis should be obtained in NB with ambigious genitalia