Congenital Diaphragmatic Hernia Congenital lung cysts Dr Baher

Congenital Diaphragmatic Hernia – Congenital lung cysts Dr Baher Sobhy

Congenital Diaphragmatic Hernia

Embryology �Normally, the intestine returns to the abdominal cavity at the 10 th week of intrauterine life, after closure of the pleuroperitoneal canal at the 8 th week. �If the closure of the pleuroperitoneal canal is delayed, the intestine will find its way to the thorax, commonly on the left side. �Accordingly, the developing lung will be compressed resulting in pulmonary hypoplasia.

Pathophysiology �The pathophysiology of CDH involves pulmonary hypoplasia and pulmonary hypertension. �The consequence of CDH is not only a reduction in the number of the alveoli, but also an increase in pulmonary resistance due to a smaller number of arterial branches with abnormal muscular hypertrophy of the media of acinar arterioles.

�This leads to pulmonary hypertension with persistence of the effect of right-left shunt through a patent ductus arteriosus. �The ductus arteriosus is going to act as a "valve" and pulmonary perfusion remain insufficient. �Persistent fetal circulation leads to a vicious cycle of progressive hypoxemia, hypercapnia and acidosis.
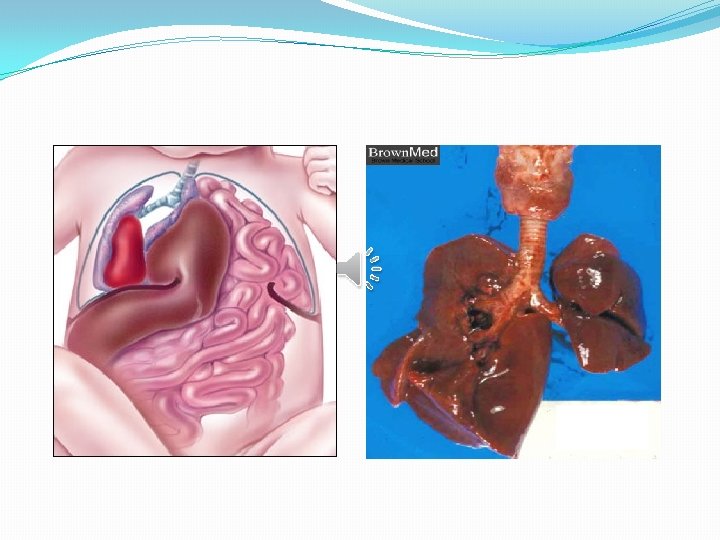

�Pulmonary hypertension may lead to right heart failure. �All resuscitation techniques implemented after birth try to get around this obstacle, by the use of pulmonary vasodilators or by the use of extracorporeal circulation (or oxygenation).

Associated anomalies � 10 -50% of patients with CDH have associated anomalies, which often contribute to the morbidity and mortality. �Most frequently associated anomalies are heart defects, chromosomal abnormalities (eg trisomy 21, 18 and 13), renal anomalies, genital abnormalities and neural tube defects.

Incidence �CDH occurs in 1 in 3000 live births. � 80% left, 20% right, rarely bilateral �Survival rate: 60 – 70%
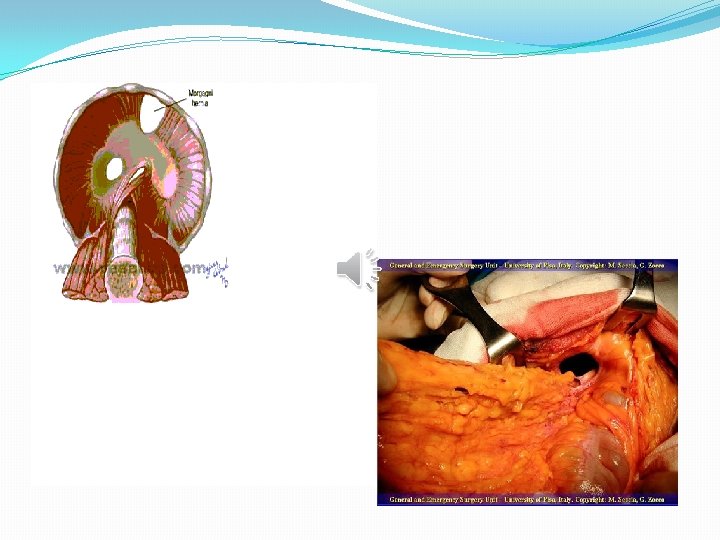
 2. Retro sternal hernia(Morgagni) 3. Eventration of diaphragm")
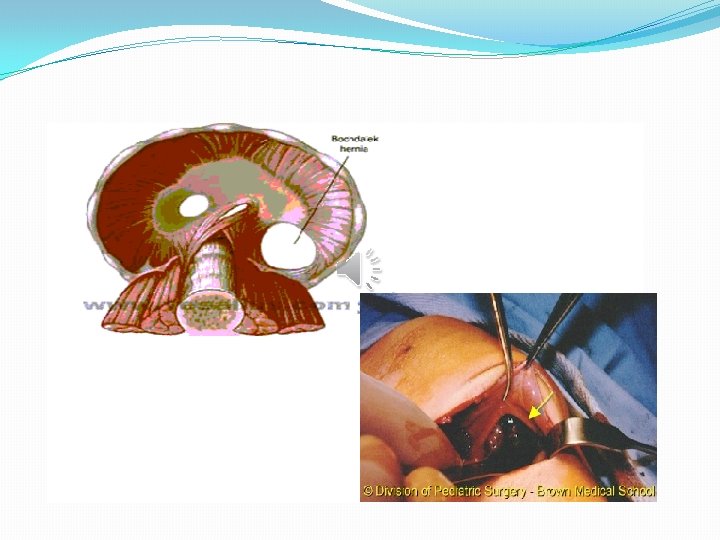
Types 1. Posterolateral hernia(Bochdalec) 2. Retro sternal hernia(Morgagni) 3. Eventration of diaphragm
, which corresponds to the")
Bochdalec hernia �Most diaphragmatic defects are posterolateral (hernia of Bochdalec), which corresponds to the persistence of the embryonic pleuro-peritoneal canal. �There may be a hemi agenesis of diaphragm. � 85 -90% of these cases occur in the left side. �A hernial sac is present in 10 -20% of cases.

Hernia of Morgagni �The hernia of Morgagni occurs posterior to the sternum and results from the failure of fusion of sternal and costal fibers where the superior epigastric artery passes through the diaphragm. �Morgagni hernia is rare, and is rarely a cause of surgery in the newborn. �A hernial sac is present in 90% of cases.

Eventration of diaphragm �Error of development of the muscular portion of the diaphragm. �Accidental diagnosis or recurrent chest infections. �Treated by plication of the diaphragm with non absorbable sutures.

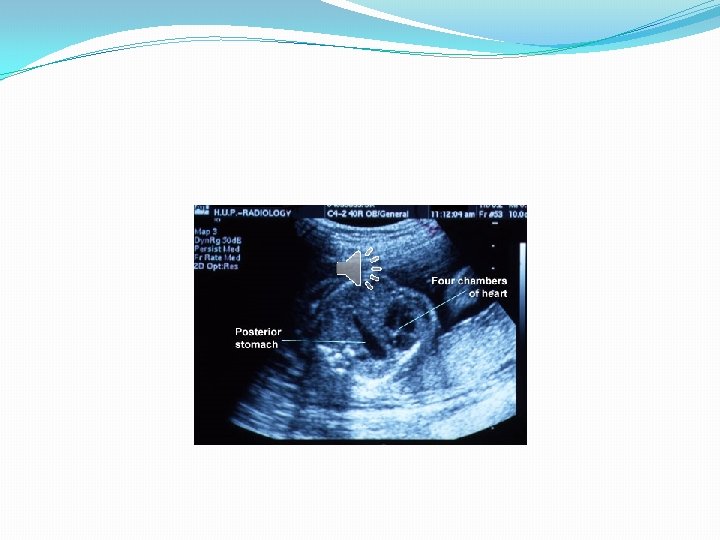
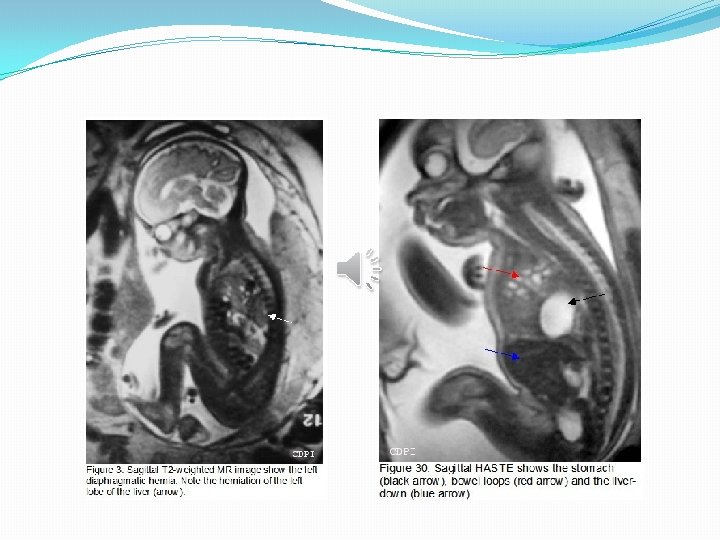
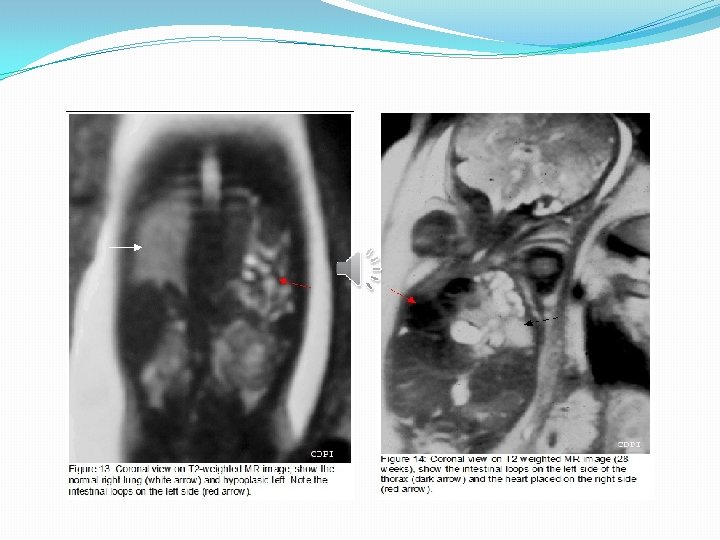
Prenatal diagnosis �The diagnosis of CDH is often prenatal from the 25 th week of gestation by ultrasound. �Recently MRI and fetal echocardiography (heart defects). �Sonographic signs include visualization of the stomach and the abdominal viscera into the thorax, presence of the heart to the opposite side of the hernia and the absence of ultrasound visualization of the diaphragm.

�Sonographic signs of severity: �Liver in the chest �Intrauterine growth retardation �fetal hydrops �Lung to head Ratio: <1 �Amniocentesis for karyotype analysis

Clinical presentation �Respiratory distress, cyanosis, tachypnea, intercostal retractions. �Newborns with large diaphragmatic defects are usually symptomatic immediately after birth, but those with small defects may be asymptomatic until several days or months of life.

�Clinical examination shows scaphoid abdomen, mediastinal shift and dispacement of cardiac sounds by auscultation. �Air entry is decreased, and bowel sounds may exist in the chest. �Forms of very late presentation are of excellent prognosis.

Investigations �Arterial blood gases. �Hypoxemia, hypercapnia and respiratory or metabolic acidosis, depend on the degree of pulmonary hypoplasia, persistent pulmonary hypertension of the newborn, the right - left Shunt, and cardiac function.

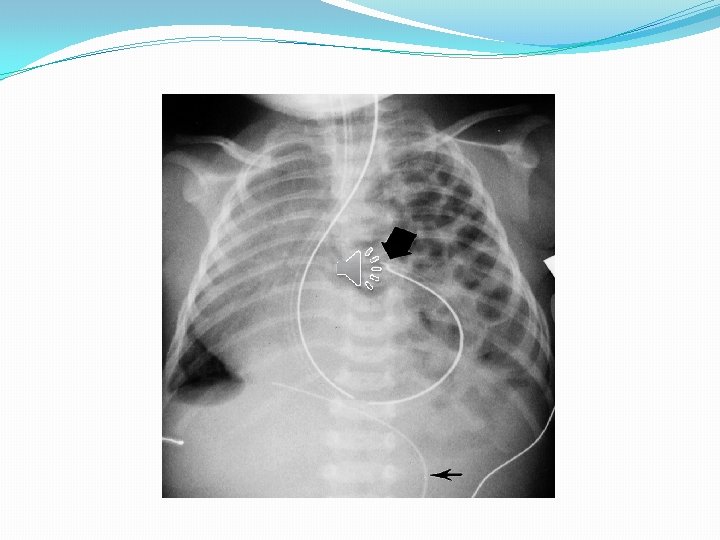
Imaging �Chest X-ray: �Intestinal loops in the chest �mediastinal shift �Absence of diaphragmatic outline �Reduction of intestinal gas in the abdomen �The presence of the tip of a nasogastric tube in the thoracic stomach.

Treatment �CDH is no longer considered as primarily a surgical disease, but is considered a disease associated with pulmonary hypoplasia, pulmonary hypertension, and pulmonary immaturity. �This led to a delayed approach to surgical repair and soft respiratory assistance.
")
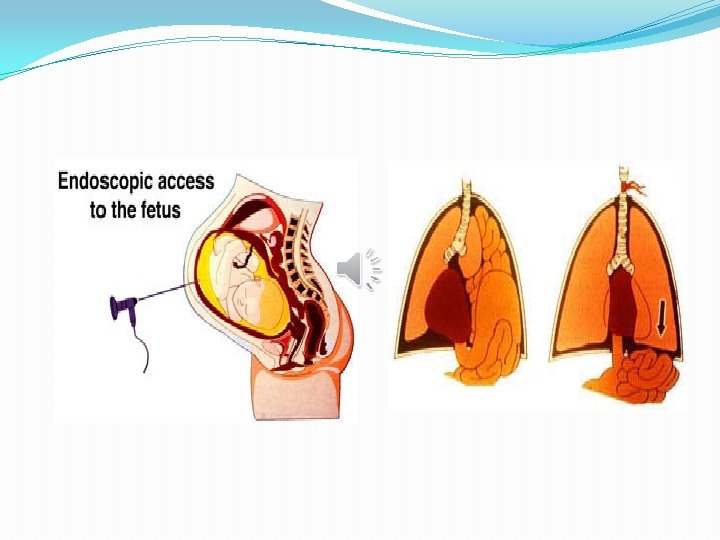
�Current research also focuses on prenatal therapies, such as in utero tracheal occlusion (FETO) to keep the secretions in the lungs and decrease pulmonary hypoplasia, by endoscopic insertion of a balloon into the trachea of the fetus, done at 26 - 28 weeks when there is ultrasonographic signs of severity with restauration of the airway before birth.

Medical treatment �Immediately after birth, the child is intubated, the use of a face mask for assisted ventilation is contraindicated to avoid gastric and intestinal distension. �A nasogastric tube is passed to decompress the stomach and prevent visceral distension.

�Proper evaluation involves continuous cardiac monitoring, measurements of arterial blood gases and blood pressure. �The limited pressure ventilation should be used. Peak inspiratory pressure (PIP) should be less than 30 cm H 2 O to prevent barotrauma to the hypoplastic lung.
 is the use of high respiration rates with low")
�High frequency oscillatory ventilation (HFOV) is the use of high respiration rates with low tidal volume to improve oxygenation of the fragile hypoplastic lung that does not tolerate conventional ventilation. �ECMO: refers to the use of extracorporeal oxygenator (artificial lung) in case of failure of maximal medical therapy.
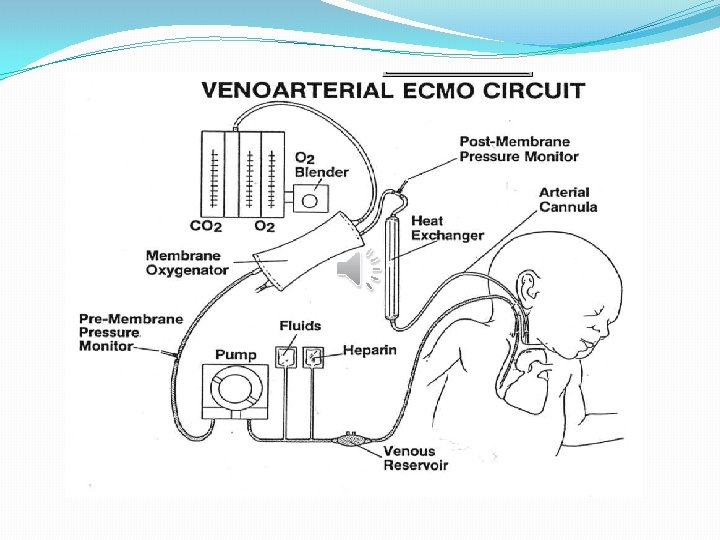
 to reduce the degree of pulmonary hypertension. �Correction of")
�Inhaled nitric oxide (pulmonary vasodilator) to reduce the degree of pulmonary hypertension. �Correction of acidosis by sodium bicarbonate. �Inotropes such as dopamine and epinephrine to improve cardiac contractility and maintain blood pressure.

Surgical treatment �Surgery is no longer considered an emergency situation, it is done after the stabilization of respiratory parameters. �There is no perfect time for the repair of CDH, but the authors suggest that the window of opportunity is 2448 hours after birth to reach normal pulmonary arterial pressure and satisfactory oxygenation and ventilation on minimal ventilator settings.

• 1. 2. 3. Aim of the operation: Reduction of the contents Closure of the defect Prevent the increase of IA pressure

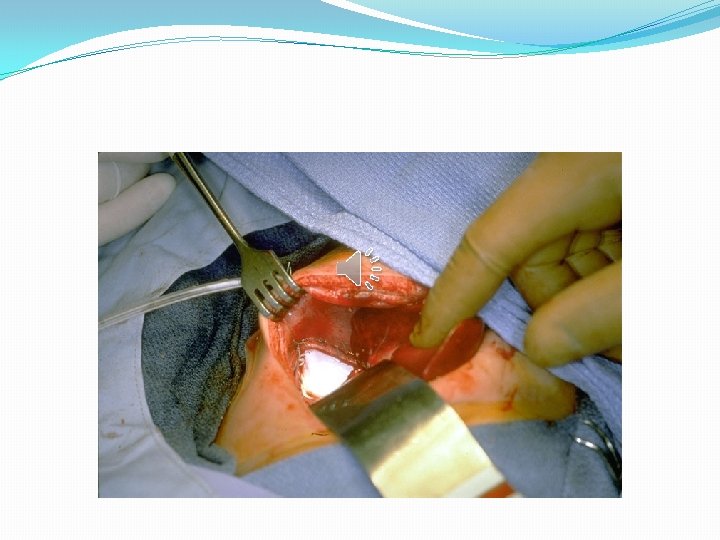
�A subcostal incision is made. Abdominal viscera are examined, and the hernia is reduced. �A hernial sac is looked for and excised if found.

�Primary repair can be accomplished in a single layer with non-absorbable sutures. �If the defect of the diaphragm is large enough to prevent the primary closure, a synthetic prosthesis can be used (gortex patch).

�Anatomical closure of the abdominal wall may not be possible after the reduction of viscera. In these cases, the skin only can be closed or a synthetic prosthesis can be used to facilitate the closure. The prosthesis can be removed with repair of the ventral hernia at a later age. �Recently, thoracoscopy or laparoscopy can be used.
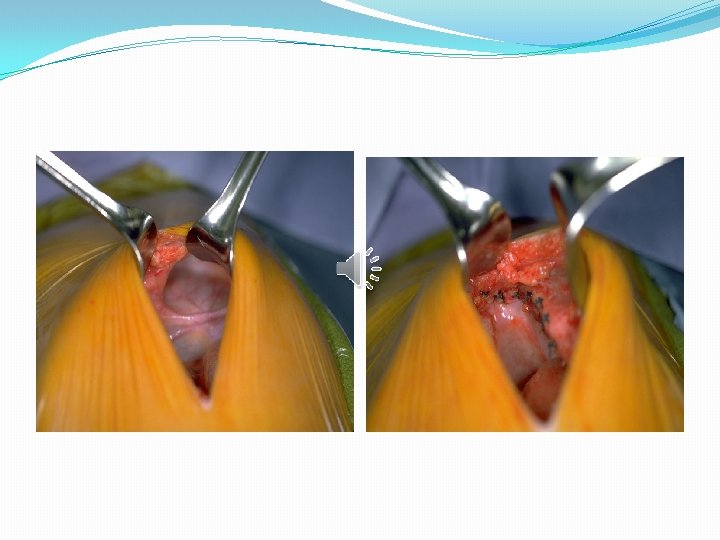

Complications �Recurrent pulmonary hypertension �Recurrence of diaphragmatic hernia �Peritoneal fluid and blood leakage in the chest, and development of an ipsilateral hydrothorax. �Bowel obstruction can be secondary to adhesions or volvulus.

Congenital lung cysts �Congenital cystic diseases of the lung are a rare but significant cause of morbidity in children and young adults presenting with respiratory distress and repeated chest infections. �They consist of: �Congenital pulmonary airway malformation (CPAM) �Bronchogenic cyst �Pulmonary sequestration �Congenital lobar emphysema
 �Formerly known as Congenital cystic adenomatoid malformations (CCAM) �Epidemiology")
Congenital pulmonary airway malformation (CPAM) �Formerly known as Congenital cystic adenomatoid malformations (CCAM) �Epidemiology �They account for about 25% of congenital lung lesions. The estimated incidence is approximately 1: 1500 -4000 live births and there is a male predominance.

Clinical presentation �The diagnosis is usually either made on antenatal ultrasound, or in the neonatal period on the investigation of progressive respiratory distress. �If large, they may cause pulmonary hypoplasia, with resultant poor prognosis. �In cases where the abnormality is small, the diagnosis may not be made for many years or even until adulthood as recurrent chest infection.

Pathology �The condition results from failure of normal bronchoalveolar development with a hamartomatous proliferation of terminal respiratory units in a gland-like pattern (adenomatoid) without proper alveolar formation. �Histologically, they are characterised by adenomatoid proliferation of bronchiole-like structures and macro- or microcysts lined by columnar or cuboidal epithelium and absence of cartilage and bronchial glands. �These lesions have intracystic communications and, unlike bronchogenic cysts, can also have a connection to the tracheobronchial tree.

Subtypes �Five subtypes are currently classified, mainly according to cyst size: �type I � most common: 70% of cases � large cysts � one or more dominant cysts: 2 -10 cm in size � may be surrounded by smaller cysts �type II � 15 -20% of cases � cysts are <2 cm in diameter � associated with other abnormalities � Renal agenesis or dysgenesis � Pulmonary sequestration � Congenital cardiac anomalies

�type III � About 10% of cases � microcysts: <5 mm in diameter � typically involves an entire lobe � has a poorer prognosis �type IV � unlined cyst � typically affects a single lobe � indistinguishable from type I on imaging �type 0 � very rare, lethal postnatally � acinar dysgenesis or dysplasia with either no cysts or very small ones � represents global arrest of lung development

�Location �Lesions are usually unilateral and involve a single lobe. Although there is no well-documented lobar predilection, they appear less frequently in the middle lobe. �Associations �Hybrid lesion: CPAM and pulmonary sequestration �Polyhydramnios �Hydrops fetalis �Lung malignancy

Radiological diagnosis �The appearance of CPAMs will vary depending on the type. �Antenatal ultrasound �CPAM appears as an isolated cystic or solid intrathoracic mass. �A solid thoracic mass is usually indicative of a type III CPAM and is typically hyperechoic. �There can be a mass effect where the heart may appear displaced to the opposite side. �The lesion may remain stable in size, or even regress. �Hydrops fetalis and polyhydramnios may develop.

�Plain radiograph �Chest radiographs in type I and II CPAMs may demonstrate a multicystic (air-filled) lesion. �Large lesions may cause a mass effect with resultant mediastinal shift, depression, and even inversion of the diaphragm. �In the early neonatal period, the cysts may be completely or partially fluid-filled, in which case the lesion may appear solid or with air-fluid levels. �Type III lesions appear solid.
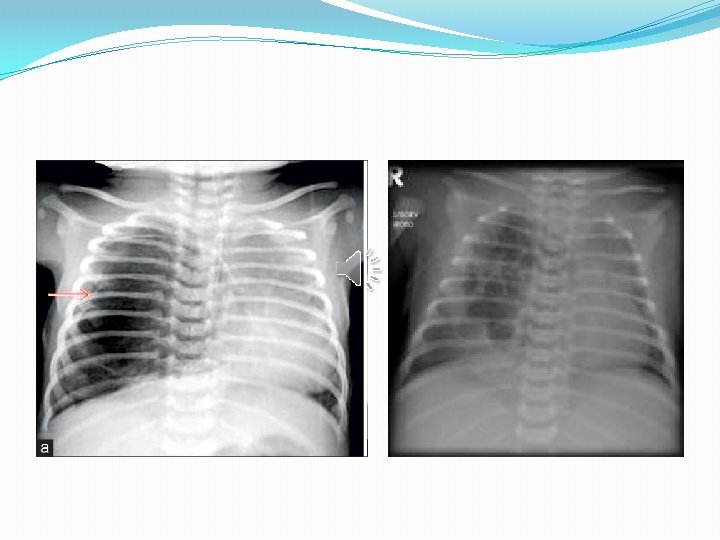

�CT �The main role of CT is to differentiate these cysts from bronchogenic cysts, lobar emphysema, and from sequestration �And to accurately localize the site of lesion within the lung segments for surgical planning.
 of variable sizes in the right upper")
� CT shows multilocular cysts (red arrow) of variable sizes in the right upper and middle lobe, and mediastinal shift to the contralateral side. �There was partial compression collapse of the right lower lobe and the left lung (green arrow).

Treatment �Treatment consists of simple resection of the involved tissue or lobectomy in symptomatic patients. �Thoracoscopy can be used. �Type I lesions have the best prognosis. �In asymptomatic lesion, surgical excision is more controversial. �Advocates for excision quote the reported risk of developing malignancies within the lesion. �An alternative approach is to watch and wait. There are reports of spontaneous regression, particularly in those serially followed up on antenatal ultrasound.

Complications �Postnatal complications include: �Pneumothorax �Pyopneumothorax �Lung malignancy �In utero complications include: �Hydrops fetalis may rarely develop when there is severe compression of the fetal heart or great vessels �Compression of the normal fetal lung can also rarely cause pulmonary hypoplasia

Bronchogenic cyst �Bronchogenic cysts arise from abnormal budding of the tracheobronchial tree during airway development. �This abnormal bud subsequently differentiates into a fluid-filled blind-end pouch. �Does not usually communicate with the bronchial tree.

�More often present in the mediastinum, one third can occur in the lung parenchyma, usually within the lower lobes. �These cysts may contain air, fluid, or both. �Clinical manifestations are related to various mass effects or secondary infection of the cyst.
 in the")
�Chest X-ray shows large thick walled air filled unilocular cyst (red arrow) in the right lung upper and mid zones. Consolidation is seen adjacent to the cyst (green arrow)

� CT chest reveals a large unilocular thick walled air filled cavity in the right upper lobe with compression of the adjacent lung parenchyma. �No obvious communication of the cyst with bronchial tree is visible
 in the")
�CT chest shows large well defined unilocular fluid containing cyst (red arrow) in the left upper lobe with thick enhancing walls. �Surrounding lung shows consolidation (green arrow)

�Complications include infection; compressive symptoms, such as dysphagia or arrhythmia; malignant transformation; and the rare but fatal air embolism. �The appropriate treatment is surgery, especially in patients with repeated infections. �Occurrence of both mediastinal and parenchymal cysts together is very rare.
 �CLE is a disease that causes breathing difficulties in newborn")
Congenital lobar emphysema (CLE) �CLE is a disease that causes breathing difficulties in newborn or infants. �Hyperluscent and hyperinflated lung segment with no cystic or solid components. �It affects left upper lobe in 43%, followed by right middle lobe in 32% of the cases. �Exact etiopathogenesis is still a dilemma, however, changes in bronchial cartilage have known to occur in most cases.

�On chest X-ray, there is over inflation of the diseased lobe resulting in shift of mediastinum to contralateral side and compression of ipsilateral lung with attenuation of vascular markings. �CT chest help to confirm these findings.
 in the left upper,")
� Chest X-ray shows a large hyperlucent area (red arrow) in the left upper, mid, and lower zone with attenuated vascular markings within the lucency (green arrow)
. �There is")
�CT shows hyperinflated left upper lobe with attenuated vascular marking (red arrow). �There is hernination of left lung field, mediastinal shift to the contralateral side with collapse of right lung (green arrow).

�CLE is commonly associated with congenital heart disease such as VSD and PDA. �Differential diagnosis includes CPAM, pulmonary hypoplasia, and pnuemothorax. �Treatment includes lobectomy of affected lung.
 is a disorder characterized by a non-functioning lung parenchyma,")
Pulmonary sequestration �Pulmonary sequestration (PS) is a disorder characterized by a non-functioning lung parenchyma, which does not communicate with tracheo-bronchial tree and receives blood supply from systemic artery. �Sequestration is believed to be due to abnormal budding of primitive foregut.

�Anatomically it can be classified as intra-lobar and extra-lobar sequestration. �Intra-lobar sequestration lies within visceral pleura and usually located in posterobasal segment of the lung. �Extra-lobar sequestration is surrounded by its own pleura, usually located on the left side of the lower chest.

�Clinically, sequestration present as �recurrent pneumonia in adults and adolescents, in case of intra-lobar �and respiratory distress, chronic cough in infancy, in case of extra-lobar sequestration. �Arterial supply is from abdominal aorta or thoracic aorta and venous drainage through pulmonary veins into left atrium. �Extra-lobar sequestration is usually associated with other congenital anomalies like diaphragmatic hernia, congenital heart disease, and CPAM.

�Radiologically sequestration on a chest X-ray is demonstrated by a soft-tissue opacity in chest base �Chest X-ray shows a homogenous dense opacity (red arrow) in right cardiophernic angle.

�CT scan show cystic areas containing air or fluid, focal emphysema, and atelectasis. �The arterial supply and venous drainage, which are pathognomic features of sequestration are better demonstrated by contrast enhanced CT scan. �Radiological differential diagnosis that need to be considered with this condition are CPAM, diaphragmatic hernia, teratoma, and accessory spleen.

Conclusion �Congenital cystic diseases of the lung are a group of lesions that share similar clinical and embryological features. �The exact incidence is not known. �Respiratory distress, cyanosis, and repeated chest infections are common symptoms. �It is important to recognize and diagnose congenital cystic lung diseases. �In view of long-term complications of infections and malignancy there is growing consensus that even asymptomatic infants should undergo elective surgery.

Thank you
- Slides: 73