COME UTILIZZARE IL DATO DELLA PDE SULLA BASE

COME UTILIZZARE IL DATO DELLA PDE SULLA BASE DELLA LINEA GUIDA SULLE SHARED FACILITIES Toni Valente Pierrel 19 aprile 2016

Evoluzione delle GMP per gli API: aspetti regolatori ed impatto sugli utilizzatori Come utilizzare il dato PDE sulla base della linea guida sulle shared facilities la rivisitazione delle attività tecniche ed organizzative alla luce degli aggiornamenti dei cap. 3 e 5 delle EU GMP in un impianto multipurpose Toni Valente Pierrel Sp. A Milano, 19 Aprile 2016

Come utilizzare il dato PDE sulla base della linea guida sulle shared facilities Il QRM per la produzione di specialità su shared facilities La valutazione tossicologica nella determinazione del livello minimo residuo di contaminante nelle Cleaning Validation Approcci alternativi al PDE

Come utilizzare il dato PDE sulla base della linea guida sulle shared facilities Il QRM per la produzione di specialità su shared facilities

• Chapter 3 e 5 pubblicati il 13 Agosto 2014 in vigore dal 1 Marzo 2015 Applicabili dal q 1 Giugno 2015 per l’introduzione di nuove specialità medicinali q 1 Dicembre 2015 per le specialità già autorizzate q 1 Giugno 2016 per le specialità veterinarie Con lo scopo principalmente di dare una guida per prevenire la cross contamination basandosi su un assessment tossicologico La nota a piè di pagina della pagina iniziale dei capitoli fa esplicito riferimento alla EMA/CHMP/CVMP/SWP/169430/2012 TOPICS CHAPTER 3 E 5

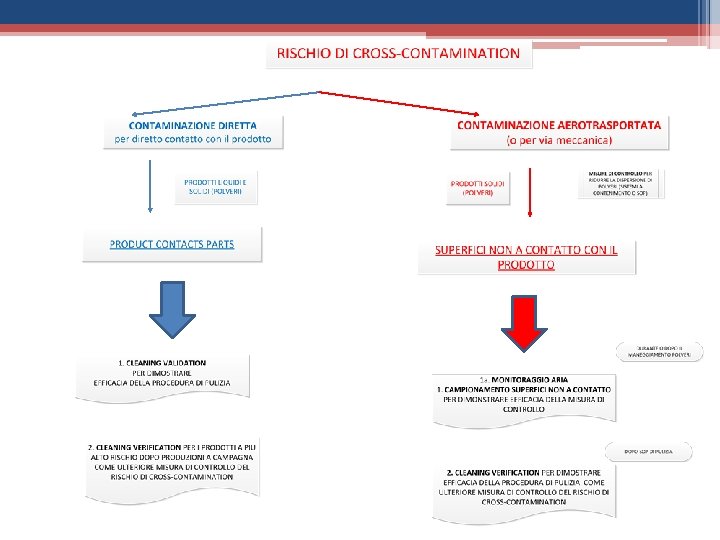
• TOPICS CHAPTER 3 E 5 Il QRM valuta il rischio di cross contamination Le dedicated facilites sono necessarie quando: 1. Il rischio non può essere contenuto con misure tecniche ed organizzative 2. La valutazione tossicologica non supporta un rischio controllabile, es. nel caso di prodotti con alto potere sensibilizzante (es. antibiotici beta-lattamici) 3. non esistono metodi analitici validati in grado di determinare il contaminante

• TOPICS CHAPTER 3 E 5 Il QRM ü Valuta e controlla il rischio di CC ANCHE sulla base di dati farmacologici e tossicologici scientificamente validi ü Determina la necessità e il livello tali per cui bisogna dedicare locali ed equipment ad un prodotto o famiglie di prodotti ü Stabilisce le misure tecniche e organizzative atte a controllare il rischio A supporto del QRM Ø La cleaning validation (annex 15/10. 6 e 10. 14) Ø La cleaning verification Ø Il campionamento delle parti NON a contatto con il prodotto per verificare l’ASSENZA di contaminazione areotrasportata o trasferita per via meccanica

• TOPICS CHAPTER 3 E 5 I risultati del QRM devono essere la base per: q Determinare le misure tecniche ed organizzative per la messa sotto controllo della cross contamination q Tali misure vanno applicate scalarmente in funzione del rischio calcolato

• TOPICS CHAPTER 3 E 5 Rischio + Misure Tecniche i. Impianti dedicati; ii. Uso di Sistemi chiusi per il trasferimento del prodotto tra gli impianti; iii. Uso di sistemi di contenimento (isolatori); iv. Parti macchina a contatto con il prodotto dedicate ; v. Uso di tecnologia monouso; vi. Uso di sistemi automatici di cleaning in place; vii. Uso di air-lock e differenziali di pressione a cascata; viii. Etc…. . Rischio -

• TOPICS CHAPTER 3 E 5 Rischio + Misure Organizzative i. Produzioni a campagne (separate nel tempo) seguite da pulizia di efficacia validata; ii. Vestizione protettiva specifica; iii. Cleaning Verification dopo ogni produzione a campagna; iv. Cleaning verification delle superfici non a contatto con il prodotto e monitoraggio dell’aria; Rischio -

• TOPICS CHAPTER 3 E 5 Chiave interpretativa dei paragrafi inseriti q Esiste il pericolo cross contamination q Tale pericolo deve essere quantificato nella sua incidenza (magnitudo) e nella sua probabilità di accadimento q A tale riguardo l’incidenza deve essere basata su dati farmacologici e tossicologici scientificamente validi (vedi PDE e/o altri approcci) q Attraverso un QRM si può definire come il pericolo lo si può controllare usando sia soluzione tecniche organizzative q Aprioristicamente nulla è incompatibile su un impianto PDE QRM

• PDE E SOSTENIBILITA’ PDE 1. il livello scientifico delle informazioni richieste (soprattutto tossicità non clinica) prevede l’intervento di un esperto delle materie citate. 2. La numerosità di API per cui è necessario il PDE genera problemi di sostenibilità da parte dei terzisti di fill e finish e dei produttori di API, che spesso non hanno un esperto interno, e che non hanno la storia tossicologica della molecola che stanno lavorando 3. I dati per calcolare il PDE non sono facilmente reperibili 4. il calcolo del limite ammesso, è fatto come se il contaminate fosse assunto per tutta la vita

• QRM: Esempi applicativi QRM q Devono essere effettuati QRM che prevedono l’introduzione di un nuovo API su una linea multi-prodotto. q Con l’ICH Q 9, le Aziende usano approcci simili, in genere è stato usato l’approccio FMEA preceduto (se necessario) da PHA o Risk Filtering. q Per le azioni di mitigazione si fa riferimento alle misure tecniche e organizzative suggerite dal punto 5. 21 del Capitolo 5. Esempio: Produzione diversi API su linea condivisa

Come utilizzare il dato PDE sulla base della linea guida sulle shared facilities La valutazione tossicologica nella determinazione del livello minimo residuo di contaminante nelle Cleaning Validation

TOPICS CHAPTER 3 E 5 Rischio + Rischio - Misure Organizzative i. Produzioni a campagne (separate nel tempo) seguite da pulizia di efficacia validata; ii. Vestizione protettiva specifica; iii. Cleaning Verification dopo ogni produzione a campagna; iv. Cleaning verification delle superfici non a contatto con il prodotto e monitoraggio dell’aria;

TOPICS CHAPTER 15 Cleaning: aspetti salienti q. La pulizia è una parte importante del processo di produzione farmaceutica q. Pulire gli impianti è un’attività non semplice! q. Poco automatizzata q. Non si possono convalidare gli operatori…. …ma possiamo misurare la loro abilità! q. Il training è fondamentale (insegnare a fare e ad essere!) q. Le procedure di pulizia devono essere riproducibili!

TOPICS CHAPTER 15 Lavare è un aspetto critico di rilevanza GMP …limits for the carryover of product residues should be based on a toxicological evaluation …prevent cross contamination… Eudralex Vol. 4 Annex 15: Qualification and Validation …The data should support a conclusion that residues have been reduced to an acceptable level… “To confirm the effectiveness of a cleaning procedure for all product contact equipment”. For all cleaning processes an assessment should be performed to determine the variable factors which influence cleaning effectiveness and performance, e. g. operators…. Particular attention should be accorded to the validation of…cleaning procedures… …Cleaning validation should be performed in order to confirm the effectiveness of a cleaning procedure… …a validation with verification after each batch may be required for some products …a visual check for cleanliness is an important part of the acceptance criteria for cleaning validation

L’approccio classico TOPICS CHAPTER 15 Il limite di accettazione definito applicando i tre seguenti criteri: • Criterio visivo • Criterio delle 10 ppm • Criterio di 1/1000 della dose Il valore più restrittivo risultante dall’applicazione dei 3 criteri definisce il livello di accettabilità dei residui di principio attivo contaminante, soddisfatto il quale è possibile dimostrare l’efficacia della procedura di pulizia. Prodotti con lo stesso dosaggio terapeutico potrebbero avere differenti profili di sicurezza API teratogeni non devono essere correlati alla dose terapeutica. Il criterio dei 10 ppm potrebbe risultare più rigoroso per farmaci a bassa attività che per farmaci ad alta attività

Nuovo approccio • TOPICS CHAPTER 1515 Alla luce di queste considerazioni, è stato necessario introdurre un approccio scientifico basato sulle attuali informazioni farmacologiche e tossicologiche del prodotto per stabilire delle soglie di accettabilità da utilizzare come parte di un risk assessment generale degli impianti multi-uso. Negli ultimi anni l'attenzione delle Autorità in materia di convalida di pulizia è cresciuta in funzione di: q Maggiore sicurezza per il paziente q «Totale validazione» (prodotto/processo) Nuovo approccio tossicologico

Normative/Linee guida in ambito Cleaning Lo stato dell’arte e l’evoluzione Ø FDA “Guide to inspections of validation of cleaning processes” 1993 Ø CEFIC/APIC (European Chemical Industry Council/Active Pharmaceutical Ingredients Committee) “Guidance on aspects of cleaning validation in active pharmaceutical ingredients plants” 2000 Ø PIC/S (Pharmaceutical Inspection Cooperation Scheme) PI 006 -03 “Recommendations on validation master plan, IQ, OQ, non-sterile process validation, cleaning validation” 2007 Ø PDA Technical Report n° 49 “Points to consider for Biotechnology Cleaning Validation” 2010 Ø ISPE “Risk-Based Manufacture of Pharmaceutical Products - A Guide to Managing Risks Associated with Cross-Contamination”, Settembre 2010 Ø PDA Technical Report n° 29 (Revised 2012) “Points to consider for Cleaning Validation” Ø EMA/CHMP/CVMP/SWP/169430/2012 “Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities”, 20/11/2014 Ø Eudralex -The rules governing medicinal products in the European Union: Volume 4, “Good Manufacturing practice - Medicinal products for human and veterinary use” a) Part 1 - Basic Requirements for Medicinal Products – Revisione 2015 Chapter 3 e Chapter 5 b) Part 2 - “Basic requirements for Active Substances used as Starting Materials” • Annex 15 Qualification and validation 2015 Ø FDA Guideline Q 3 D Elemental Impurities Settembre 2015

L’evoluzione normativa ISPE Baseline Guide® Risk-Ma. PP Ø La linea guida ISPE®Risk-Mapp fornisce un approccio scientifico basato sul rischio, in linea con la linea guida ICH Q 9, per gestire il rischio di contaminazione crociata nella produzione di principi attivi farmaceutici e prodotti farmaceutici. Ø Nell'ambito del Risk-Mapp, viene proposta una valutazione del rischio basata sulla sicurezza del paziente e scientificamente sostenibile allo scopo di stabilire l’Acceptable Daily Exposure (ADE). Ø L'ADE può essere utilizzata come base per calcolare la quantità massima di API o altro materiale che può essere presente nel prodotto successivo. Ø L'ADE rappresenta una stima dell’esposizione quotidiana ad una sostanza che non presenta rischi apprezzabili di effetti dannosi per la popolazione potenziale di pazienti durante un tempo di vita medio. Ø Il concetto di ADE è simile ai limiti sviluppati da altri enti regolatori così come dai singoli produttori per definire i limiti "sicuri" o "accettabili". 22
: La dose massima")
L’evoluzione normativa ISPE Baseline Guide® Risk-Ma. PP Acceptable Daily Exposure (ADE): La dose massima del prodotto che un individuo può assumere tutti i giorni senza probabili effetti avversi ADE (mg/day) = NOAEL (mg/kg/day) x BW (kg) UFC x MF x PK NOAEL = No Observed Adverse Effect Level BW = peso corporeo (es. kg 50) UFC = Composite Uncertainty Factor (Intraspecies Differences, Interspecies Differences, Subchronic -to-Chronic Extrapolation, LOAEL-to-NOAEL Extrapolation, Database Completeness) MF = Modifying Factor (fattore basato sul giudizio del tossicologo) PK = Pharmacokinetic factor (es. route-to-route extrapolation) Nell’ambito del risk assessment, combinato con la valutazione dell’esposizione del prodotto, l’ADE permette il calcolo quantitativo del rischio associato ad ogni situazione di pericolo individuata. Inoltre, l’ADE viene utilizzato come base per il calcolo del limite di residuo accettabile nelle procedure di cleaning 23
 “Points to consider for Cleaning")
L’evoluzione normativa PDA Technical Report n° 29 (Revised 2012) “Points to consider for Cleaning Validation” Principi attivi «altamente pericolosi» Risk Ma. PP Acceptable Daily Exposure (ADE) Principi attivi non «altamente pericolosi» Risk Ma. PP Acceptable Daily Exposure (ADE) o criterio basato sulla dose terapeutica Sostanze che non hanno una dose terapeutica criterio basato sulla LD 50 24

TOPICS CHAPTER 15

TOPICS CHAPTER 15 EMA/CHMP/ CVMP/ SWP/169430/2012 - goal Applicata a tutti i prodotti medicinali per uso umano e veterinario (anche prodotti in fase di sperimentazione e API. . ) prodotti in locali adibiti anche per la fabbricazione di altri medicinali. Il punto finale è quello di garantire beneficio per il paziente, ovvero non creare danno Tuttavia un contaminante non fornisce beneficio per il paziente e può anche rappresentare un rischio. La presenza di tali contaminanti deve essere gestita secondo il rischio relativo ai livelli che possono essere considerati sicuri per tutte le popolazioni. Limiti di sicurezza (Health based limits) correlati a valori di soglia di sicurezza (safe threshold value) devono essere impiegati per identificare i rischi.

EMA/CHMP/ CVMP/ SWP/169430/2012 - goal Categorizzazione delle sostanze attive utilizzate nelle proprie formulazioni secondo i Regolamenti REACH/CLP, al fine di poter classificare correttamente gli API secondo le seguenti caratteristiche: q. Genotossiche q. Tossicità riproduttiva e sullo sviluppo q. Carcinogene q. Altamente sensibilizzanti

EMA/CHMP/ CVMP/ SWP/169430/2012 - considerazioni q Il PDE è calcolato con la formula proposta da ICH Q 3 C (R 5). q Q 3 C basa il calcolo tenendo conto del NOEL (No Observed Effect Level). q Nella versione approvata il PDE è in funzione del NOAEL (No Observed Adverse Effect Level). q Considerata la correlazione concettuale tra NOEL, NOAEL, LOAEL, come indicata dal grafico, il NOEL rappresenta un valore più piccolo rispetto al NOAEL per il calcolo del PDE
: PDE (mg/day) = NOAEL (mg/kg/day)")
EMA/CHMP/ CVMP/ SWP/169430/2012 - considerazioni Permitted Daily Exposure (PDE): PDE (mg/day) = NOAEL (mg/kg/day) x BW (kg) F 1 x F 2 x F 3 x F 4 x F 5 F 1: un fattore (tra 2 e 12) che tiene conto dell’estrapolazione tra le specie F 2: un fattore di 10 che tiene conto della variabilità tra individui F 3: un fattore di 10 che tiene conto di studi di tossicità di breve durata con dosi ripetute es. , meno di 4 settimane F 4: un fattore (1 -10) che può essere applicato in caso di tossicità severa, e. g. carcinogenicità non-genotossica, neurotossicità o teratogenicità F 5: un fattore variabile che può essere applicato se non è stato stabilito nessun effetto. Quando è disponibile solo il LOEL, può essere utilizzato un fattore fino a 10 a seconda della severità della tossicità. 29
: EMA Acceptable Daily Exposure (ADE):")
EMA/CHMP/ CVMP/ SWP/169430/2012 - considerazioni Permitted Daily Exposure (PDE): EMA Acceptable Daily Exposure (ADE): ISPE Risk Ma. PP, FDA EC GMP Vol. 4 Annex 15 – Qualification and Validation, 30/03/2015: 10. 6 Limits for the carryover of product residues should be based on a toxicological evaluation (see EMA Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities). The justification for the selected limits should be documented in a risk assessment which includes all the supporting references. L’APPROCCIO EMA E’ PIU’ CONSISTENTE CON QUELLO ISPE Risk-Ma. PP (Risk-based Manufacture of Pharmaceutical Products)! 30

EMA/CHMP/ CVMP/ SWP/169430/2012 - considerazioni Adoperando il NOAEL piuttosto che il NOEL, EMA and Risk-Ma. PP usano le stesse basi per determinare il livello di sicurezza. …. “PDE and ADE are effectively synonymous”…. . Ref. : How Clean is Clean in Drug Manufacturing? The review of American Pharmaceutical Business & Technology - August 25, 2014

APPROCCIO DI CALCOLO NOEL Linea Guida APIC "Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants" NOEL = LD 50 x BW / 2000 dove: • NOEL: dose senza effetto avverso osservabile (No Observed Effect Level). • LD 50: Dose Letale 50 in mg/Kg animale. L’identificazione dell’animale (topo, ratto etc. ) e la modalità di somministrazione (IV, oral etc. ) è importante (mg/kg). • BW: E 'il peso medio di un adulto. In questo caso si accetta il valore di 50 Kg • 2000: È una costante empirica I valori della LD 50 sono stati estrapolati da studi di tossicità acuta condotti su animali da laboratorio e formalizzati in articoli di letteratura che si possono anche trovare nelle pertinenti sezioni del Dossier di Registrazione di un farmaco (Modulo 4 – Pre clinical Study Reports). Moltiplicando il valore di LD 50 per il valore del peso corporeo di un adulto e di un bambino e dividendo il prodotto per 2000, si ricava il valore di NOEL. L’approccio di sostituire il valore di NOAEL con il parametro NOEL, permette di fissare il fattore F 5 uguale ad 1.

Integrazione del tool tossicologico nel QRM q Risk Assessment ü layout stabilimenti produttivi ü prodotto ü processo produttivo ü procedure di pulizia ü flussi materiale e personale q Risk Mitigation ü Attuazione delle misure di risk mitigation (es. dedicated facilities/equipments)

Esempi applicativi QRM: Produzione su Linea condivisa con altri API

Esempi applicativi QRM: Produzione su Linea condivisa con altri API - MODO DI GUASTO (PERICOLO) PROBABILITA' [1 -5] Cross Contamination chimica aerotrasportata tra materie prime Cappe a contenimento a flusso laminare non funzionanti o spente MOLTO ALTO 5 BASSO Controllo periodico 2 MEDIO controllo secondo SOP 3 Inadeguato Non corretta applicazione Cross-contamination delle della procedura di Flusso dei Materiali maneggiamento delle materie prime Materie Prime dispensazione MOLTO ALTO 5 BASSO Personale trainato 2 MEDIO controllo secondo SOP Cross Contamination Non corretta applicazuione chimica aerotrasportata della procedura di tra materie prime vestizione MOLTO ALTO 5 BASSO Personale trainato; Vestizione monouso per singola materia prima 2 MEDIO Tools per la dispensazione con residui di polveri MOLTO ALTO 5 BASSO Tools monouso sterili per singola materia prima 2 MEDIO Flusso del Personale Vestizione inadeguata del Personale Cleaning Process Procedura di pulizia non applicata corretamente per le superfici a contatto Cleaning Process Mancata o incompleta Pulizia di Aree e Superfici Cross-contamination Cross Contamination chimica aerotrasportata tra materie prime Non corretta applicazione della procedura di pulizia MOLTO ALTO G RILEVABILITA' [1 -5] POTENZIALE CAUSA Dispersione Polveri in Equipments/Facilit ambiente del locale y dispensing /sampling DISPENSING/SAMPLING SEVERITA' (GRAVITA') [1 -5] EFFETTO DEL GUASTO 5 BASSO Personale trainato P 2 MEDIO R VALORI RPN AZIONE CORRETTIVA 0 na 64 azione immediata RISCHIO High Risk Threshold Value= (4 x 4 x 4) RPN [1 -125] AZIONI CORRETTIVE NOTE / SUGGERIMENTI 30 na Monitoraggio aria e campionamento delle superfici non a contatto con il prodotto secondo quanto prescritto dal capitolo 5 delle GMP al fine di verificare il grado di contaminazione aereotrasportata e l'efficacia della misura di controllo presente (cappa a contenimento polveri). 3 30 na - controllo secondo SOP 3 30 na - na Una volta assodato il grado/livello di contaminazione aerotraportata o tarsferita per via meccanica delle superfici non a contatto con il prodotto, Cleaning Validation/Verification per verificare l'efficacia della procedura di pulizia come ultima misura di controllo/mitigazionedel rischio di crosscontamination (prima del lotto successivo). 3 controllo secondo SOP SEVERITA' PROBABILITA’ MACROFASE 1 2 3 5 5 20 45 4 4 16 36 3 3 12 27 2 2 8 18 1 1 4 9 1 2 3 NON RILEVABILITA’ 4 5 80 125 64 100 48 75 32 50 16 25 4 5 30

Esempi applicativi QRM: Produzione su Linea condivisa con altri API - MODO DI GUASTO (PERICOLO) SEVERITA' (GRAVITA') [1 -5] POTENZIALE CAUSA PROBABILITA' [1 -5] G RILEVABILITA' [1 -5] P R RPN [1 -125] AZIONI CORRETTIVE NOTE / SUGGERIMENTI MOLTO ALTO Sistema di monitoraggio in continuo dei Delta. P con allarmi visivi e sonori 1 10 na Monitoraggio aria e campionamento delle superfici non a contatto con il prodotto secondo quanto prescritto dal capitolo 5 delle GMP al fine di verificare il grado di contaminazione aereotrasportata e l'efficacia della misura di controllo presente (LAF/SOP Flusso dei Materiali). MOLTO ALTO 5 BASSO Controllo periodico; travaso diretto delle materie prime nel dissolutore Inadeguato Non corretta applicazione Cross-contamination delle della procedura di Flusso dei Materiali maneggiamento delle materie prime Materie Prime preparazione MOLTO ALTO 5 BASSO Personale trainato; travaso diretto delle materie prime nel dissolutore sotto LAF 2 MEDIO controllo secondo SOP 3 30 na - Cross Contamination Non corretta applicazuione chimica aerotrasportata della procedura di tra materie prime vestizione MOLTO ALTO 5 BASSO Personale trainato; Vestizione monouso per singolo lotto 2 MEDIO controllo secondo SOP 3 30 na - Dispersione Polveri in Equipments/Facilit ambiente del locale y compounding PREPARAZIONE EFFETTO DEL GUASTO Flusso del Personale Vestizione inadeguata del Personale Cross Contamination chimica aerotrasportata tra materie prime Efficienza HVAC / LAF 2 Cleaning Process Mancata o incompleta Pulizia di Aree e Superfici Cross Contamination chimica aerotrasportata tra materie prime Non corretta applicazione della procedura di pulizia MOLTO ALTO 5 BASSO Personale trainato 2 MEDIO controllo secondo SOP 3 30 na Una volta assodato il grado/livello di contaminazione aerotraportata o tarsferita per via meccanica delle superfici non a contatto con il prodotto, Cleaning Validation/Verification per verificare l'efficacia della procedura di pulizia come ultima misura di controllo/mitigazionedel rischio di crosscontamination (prima del lotto successivo). Cleaning Process CIP e SIP non eseguiti correttamente Cross-contamination errore umano nell'applicazione della procedura; MOLTO ALTO 5 MOLTO BASSO Processo automatizzato 1 MEDIO controllo secondo SOP 3 15 na Cleaning Verification sulle acque di rinsing per i prodotti a più alto rischio dopo produzioni a campagna come ulteriore misura di controllo del rischio di cross-contamination. SEVERITA' VALORI RPN AZIONE CORRETTIVA 0 na 64 azione immediata RISCHIO High Risk Threshold Value= (4 x 4 x 4) PROBABILITA’ MACROFASE 1 2 3 5 5 20 45 4 4 16 36 3 3 12 27 2 2 8 18 1 1 4 9 1 2 3 NON RILEVABILITA’ 4 5 80 125 64 100 48 75 32 50 16 25 4 5

Esempi applicativi QRM: Produzione su Linea condivisa con altri API - MODO DI GUASTO (PERICOLO) EFFETTO DEL GUASTO POTENZIALE CAUSA SEVERITA' (GRAVITA') [1 -5] G Flusso dei Materiali Inadeguato maneggiamento delle parti macchina a contatto con il prodotto Cross-contamination chimica Non corretta applicazione della procedura di flusso dei materiali MOLTO ALTO 5 BASSO Personale trainato 2 MEDIO Cleaning Process Procedura di Lavaggio manuale delle parti macchina/riempitrice non eseguita correttamente Cross-contamination chimica errore umano nell'applicazione della procedura; MOLTO ALTO 5 BASSO Personale trainato 2 MEDIO PROBABILITA' [1 -5] RILEVABILITA' [1 -5] P R RPN [1 -125] AZIONI CORRETTIVE NOTE / SUGGERIMENTI controllo secondo SOP 3 30 na - controllo secondo SOP 3 30 na Cleaning Verification sulle acque di rinsing per i prodotti a più alto rischio dopo produzioni a campagna come ulteriore misura di controllo del rischio di cross-contamination. SEVERITA' VALORI RPN AZIONE CORRETTIVA 0 na 64 azione immediata RISCHIO High Risk Threshold Value= (4 x 4 x 4) PROBABILITA’ LAVAGGIORIEMPIMENTO MACROFASE 1 2 3 5 5 20 45 4 4 16 36 3 3 12 27 2 2 8 18 1 1 4 9 1 2 3 NON RILEVABILITA’ 4 5 80 125 64 100 48 75 32 50 16 25 4 5

EMA/CHMP/ CVMP/ SWP/169430/2012 - considerazioni Criterio tossicologico: L = PDE x DLT x 106 DMAB x S L: limite di accettabilità (μg/cm 2) PDE: Permitted Daily Exposure (mg/die) DLT: dimensione del lotto del prodotto contaminabile (kg) DMAB: dose massima giornaliera del prodotto contaminabile (g/die) S: area delle superfici comuni delle attrezzature coinvolte (cm 2) L’estremizzazione di safety di questo approccio è che si ipotizza che una sostanza A contamini sempre tutti i lotti di una specialità B e che questa venga somministrata per tutta la vita ad un paziente L’organizzazione industriale difficilmente o raramente prevede questo approccio 45

EMA/CHMP/ CVMP/ SWP/169430/2012 - considerazioni Worst case Severità Valori corrispondenti PDE mg/kg/giorno 1 >15 2 10≤ PDE≤ 15 3 5≤ PDE≤ 10 4 2≤ PDE≤ 5 5 <2 probabilità solubilità 1 Altamente solubili 2 Solubili con matrice insolubile 3 Poco solubili con matrice poco solubile 4 Insolubili e con matrice poco insolubile 5 Insolubili con matrice insolubili Rilevabilità Valori corrispondenti 1 Limite di accettabilità ≥ al criterio visivo (4, 0 μg/cm 2) 2 2, 0 μg/cm 2≤ limite d accettabilità≤ 4, 0 μg/cm 2 3 1, 5 μg/cm 2≤ limite d accettabilità≤ 2, 0 μg/cm 2 4 1, 0 μg/cm 2≤ limite d accettabilità≤ 1, 5 μg/cm 2 5 Limite di accettabilità <1, 0 μg/cm 2 46

Come utilizzare il dato PDE sulla base della linea guida sulle shared facilities Approcci alternativi al PDE
")
Guideline: topics 1. Possibilità di metodologie alternative al calcolo del PDE (introduction, par 1) 2. Possibilità di usare un approccio basato sui dati clinici (non PDE based) se il principale effetto critico è osservato sull’uomo piuttosto che sull’animale (par 4. 2) 3. Classificare il livello di sensibilizzazione delle sostanze sulla base della frequenza e della gravità delle reazioni sull’uomo, OPPURE su dati animali o altri test validati (par 5. 2) 4. Per gli IMP nelle prime fasi di sviluppo e con pochi dati tossicologici a disposizione possono essere usati approcci alternativi Nella presentazione il termine PDE è riferito all’applicazione della linea guida MA L’approccio AFI è un metodo alternativo per calcolare il limite di assunzione giornaliera considerato sicuro, in pratica è un altro modo per calcolare il Permitted Daily Exposure
 2. Le valutazioni in base")
CRITICITA’ 1. Richiede l’intervento di un esperto (es. tossicologo) 2. Le valutazioni in base a “letteratura di qualità”: NON è standard, può non essere approvata dall’autorità regolatoria, può essere diversa per lo stesso API. 3. NON è proposta una metodologia oggettiva (es. tools usati nell’analisi del rischio) per analizzare la letteratura 4. La metodologia proposta può generare BIAS, tutto è demandato al giudizio dell’esperto incluso il reperimento della “letteratura di qualità” Sulla base di tali evidenze è costruito un approccio alternativo

Obiettivi Ø Approccio sostenibile Ø Metodologia di analisi STANDARD, OGGETTIVA E USANDO FONTI ACCREDITATE DALL’AUTORITA’ REGOLATORIA ü Esprime una valutazione tossicologica tramite l’analisi dei dati in RCP (con particolare attenzione ai dati sull’uomo), via Risk Filtering ü ALTERNATIVA al PDE: particolarmente per farmaci di uso terapeutico consolidato ü PDE: approccio preferito per molecole nuove (PDE nella MAH), limitata disponibilità di dati sull’uomo, particolari criticità

La QP Chi valuta: competenze e basi normative Come da sempre è responsabilità finale della QP garantire - tramite un team multidisciplinare - che il livello di contaminate considerato accettabile sia sicuro per il paziente, a prescindere dalla metodologia con cui è calcolato o da chi lo calcola. In base all’art 49 della 2001/83 e all’art 52 del 219/2006 viene conferita l’idoneità QP solo se il richiedente è in possesso di “formazione a livello universitario deve comprendere gli insegnamenti teorici e pratici delle seguenti discipline di base e il superamento dei relativi esami: fisica sperimentale, chimica generale ed inorganica, chimica farmaceutica, compresa l'analisi dei medicinali, biochimica generale e applicata, fisiologia, microbiologia, farmacologia, tecnologia farmaceutica, tossicologia, farmacognosia” analitica, Sulla base di tali competenze la QP è ESPERTO nel valutare il livello sicuro di farmaco da considerare nelle analisi del rischio e nella cross contamination.

Fonte dei dati: RCP Valore legale: l’AIC è pubblicata in Gazzetta Ufficiale • Parte integrale dell’AIC che ogni AR deve valutare EMA O O T A V APPRO ORITA’ AUT TORIA A L O REG • Ogni modifica: approvata dall’AR Aggiornato e completo tutte le informazioni del farmaco, PRIMA e DOPO la sua approvazione

PRIMA Molti esperti studiano il farmaco Aspetti tossicologici sono valutati negli studi preregistrativi conformi alle ICH MA O ATO E V O R APP RITA’ AUTO ORIA LAT REGO DOPO Monitoraggio continuo, EMA e Tit AIC Dal 1° settembre 2015 EMA ha attivato il servizio di monitoraggio della letteratura medica, e il relativo inserimento delle reazioni avverse in Eudra. Vigilance, esteso a 100 medicinali a base di erbe e 300 gruppi di sostanze chimiche inclusi nell'elenco dell’EMA. …l'EMA effettua un monitoraggio sistematico di una selezione della letteratura medica per individuare le segnalazioni di sospette reazioni avverse contenenti determinate sostanze attive. E' anche previsto che l'EMA provveda a registrare i casi individuali nella banca dati Eudravigilance…. I titolari dell’AIC ……, sono comunque tenuti a monitorare tutta la restante letteratura medica e a trasmettere qualsiasi sospetta reazione avversa…. . AO TO EM A V O APPR RITA’ AUTO ORIA LAT REGO
 oltre")
Come valuta: metodologia Per ogni farmaco si esegue un Risk filtering (6 fattori) oltre l’RPN è consigliato il calcolo del PDE; sotto l’RPN si usa come base di calcolo la dose minima terapeutica da RCP/1000 1. benchmark dei limiti applicati con successo da circa 20 anni a ora 2. Molto cautelativo, integrato con una valutazione tossicologica 3. Base dei dati storici aziendali e del PQS, da usare nelle valutazioni del rischio Dose minima terapeutica: facilmente e univocamente reperibile dall’RCP. Ø Il Risk Filtering classifica il farmaco: è un’analisi riferita unicamente all’API (PERICOLO), formulato secondo una via di somministrazione che deve corrispondere a quella degli altri farmaci prodotti sulla stessa linea (es. se ho i dati per un uso topico non vanno bene se sulla stessa linea produco orali) Ø tutte le altre variabili tecniche e organizzative vengono valutati nell’esercizio di QRM (RISCHIO).

Topics della linea guida fatt NO corr TE SCORE (se mancano i dati inserire cautelativamente 5) FATTORE, par RCP POTENZA API-CHIMICO 1 (mg/die) POTENZA API-BIOLOGICO EFFETTO CRITICO GRAVITA' RCP: 4. 8, 4. 3, 4. 4, 4, 5 2 EFFETTO CRITICO PROBABILITA‘ RCP: 4. 8, 4. 3, 4. 4, 4, 5 3 FERTILITA’, GRAVIDANZA, ALLATTAMENTO RCP: 4, 6 4 5 6 USO PEDIATRICO RCP 4. 3 ANNI DI COMMERCIALIZZAZIONE ESTENSIONE DEL MERCATO (ALMENO 1 TRA I PAESI DELLE AREE ICH) 1 2 3 4 5 ≥ 1000 ≥ 100 -1000 ≤ ≥ 10 - 100 ≤ ≥ 1 - 10 ≤ 1≤ n/a TBD TBD TBD n/a 1< 1 2 3 4 -5 0, 5 B very rare (<1/10, 000) rare (≥ 1/10, 000 to <1/1, 000) uncommon (≥ 1/1, 000 to <1/100) Common (≥ 1/100 to <1/10) Uso consentito n/a ≥ 20 ≥ 10 - 20 ≤ ≥ 5 - 10 ≤ ≥ 1 -5≤ EU, Japan, Nord America 2 Almeno 1 n/a Very common 0, 5 (≥ 1/10) vietato controindicato perché per mancanza di dati esistono N/A dati sull’uomo e sull’uomo, disponibili dati sull’animale sull’uomo informazioni non controind n/a disponibili icato 1≤ n/a NESSUNO n/a TOTALE A) dose minima terapeutica giornaliera; associazioni: la somma di tutti B) Specificare quale/i effetti gravi sono stati scelti e perché C) Specificare a quale probabilità si fa riferimento A C

GIUSTIFICAZIONE DELL’RPN Valori 1 -30; RPN = 18; per valori > 18 è consigliato il calcolo del PDE fattore CONTRIBUTO PER RPN potenza 3 gravità 3 (x 0. 5 ) frequenza 3 (x 0. 5) Fertilità gravidanza allattamento 3 Anni di commercializzazione 3 Perché il prodotto ha subito almeno un rinnovo quinquennale, inoltre ha superato i primi anni di commercializzazione in cui spesso c’è monitoraggio intensivo Uso pediatrico 3 Perché tre corrisponde ad una NON controindicazione del prodotto per l’uso in pediatria, molti farmaci sono normalmente usati su popolazioni pediatriche pur in assenza di studi clinici specifici sulla popolazione in oggetto. 3 Perché rispetto al totale dei paesi ICH , che corrisponde a valore 1, e a nessun paese ICH, che corrisponde a valore 5, il valore 3 numericamente esprime una media dell’estensione dei mercati che comunque è adeguata per monitorare eventuali effetti critici Estensione dei mercati motivazione Perché 10 -100 mg/die è cautelativo in quanto non identifica il 50% della dose con fattore di rischio 1 ma un quantitativo inferiore Perché corrisponde al livello CTCAE (2) che ha effetti lievi sul paziente e non lo mettono a rischio di vita Perché la frequenza esprime un valore numerico che non è possibile valutare qualitativamente e quindi si considera la media L’uso è controindicato però mancano i dati sull’uomo e sull’animale, quindi è una controindicazione che non emerge da una diretta evidenza di tossicità sull’uomo nelle condizioni considerate. Per una valutazione completa di ogni fattore è importante leggere TUTTI i paragrafi dell’RCP, non solo quelli di principale riferimento

EFFETTO CRITICOGRAVITA’ EFFETTO CRITICOPROBABILITA’’

Valutazione farmaceutica Vs valutazione tossicologica • EFFETTO GRAVE • A DOSAGGI TERAPEUTICI • PROLUNGATI PER ANNI t case) eri I num ors (es. w o n a t con Numero mg/die (o mg/Kg) PIU’ ALTO Riflette un pericolo che realmente possiamo portare al paziente • EFFETTO MENO GRAVE (COMUNQUE SERIO) • DOSAGGI BASSI O DOSE INDIPENDENTE • SOMMINISTRAZIONI PER BREVI PERIODI O SINGOLE Numero mg/die o mg/Kg PIU’ BASSO ? Riflette un pericolo che possiamo portare al paziente SI La scelta dell’effetto critico richiede una valutazione farmaceutica, non solo tossicologica: bisogna motivare il limite, non considerare solo il più basso ma considerare quello che protegge meglio il paziente TOSSICOLOGIA VALUTAZIONE TOSSICOLOGICA 219/2006 ……“…fisica sperimentale, chimica generale ed inorganica, chimica analitica, chimica farmaceutica, compresa l'analisi dei medicinali, biochimica generale e applicata, fisiologia, microbiologia, farmacologia, tecnologia farmaceutica, TOSSICOLOGIA, farmacognosia” VALUTAZIONE FARMACEUTICA
 ICH Q 3 C solventi APPROCCIO AFI PDE (guideline)")
APPROCCIO AFI Vs PDE (guideline) ICH Q 3 C solventi APPROCCIO AFI PDE (guideline) Fonte dei dati validata dall’autorità regolatoria SI Reperibilità dei dati FACILE COMPLESSA Soggettività della valutazione BASSA ALTA Sostenibilità, complessità tecnica ed economica, e applicabilità FACILE DIFFICILE Tempestiva, dopo valutazione Non programmabile Aggiornamento dati: tempestività e periodicità Integrazione con il PQS Interpretabilità da parte del personale coinvolto nella attività farmaceutiche (QA, auditor) EMA o Autorità Regolatoria NO (non obbligatoria) dell’RCP è immediato BASSA, revisioni periodiche? ALTA BASSA ALTA, l’aggiornamento

Conclusioni üValutazione tossicologica che mitiga alcune criticità dell’approccio PDE üSe si applica l’approccio della guideline: pre-analisi che allinea le valutazioni del tossicologo e della QP üE’ la QP che decide quale approccio usare: non solo perché è il responsabile finale ma perché ha tutte le competenze per valutare i dati üE’ la QP che decide se e quali esperti coinvolgere: es. esperti nell’effetto considerato critico, approccio misto AFI è una Società Scientifica Base di partenza: AFI stimola i soci a dare il proprio contributo per migliorare l’ approccio.

Question time

Ogni riferimento a persone esistenti o fatti realmente accaduti è puramente casuale, specie quando si parla di …………. . cross contamination

Grazie per l’attenzione
- Slides: 56