COLLAGEN METABOLISM Collagen is the most abundant protein

COLLAGEN METABOLISM

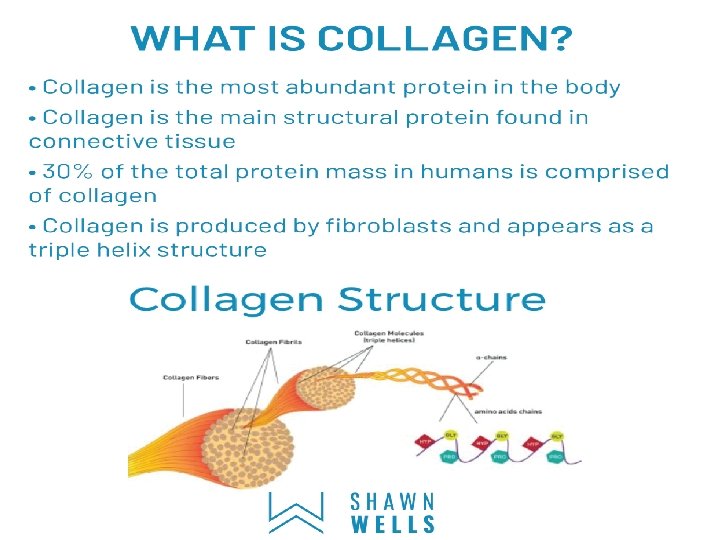
Collagen is the most abundant protein in the human body. The typical collagen molecule is a long, rigid structure in which three polypeptides (referred to as α chains) are wound around one another in a rope-like triple helix. collagen is agel like that gives support to the structure, as in the extracellular matrix or the vitreous humor of the eye. In other tissues, collagen may be bundled in tight, parallel fibers that provide great strength, as in tendons. In the cornea of the eye, collagen is stacked so as to transmit light with a minimum of scattering. Collagen of bone occurs as fibers arranged at an angle to each other so as to resist mechanical shear from any direction.
")
Structure of collagen Collagen is made up of three polypeptides (referred to as "α-chains") are twisted around one another (tropocollagen) in a rope-like triple-helix and are held together by hydrogen bonds. Collagen is formed from tropocollagen subunits. The triple helix in tropocollagen is highly extended and strong. Features: (1) Three separate polypeptide chains arranged as a left-handed helix (note that an alpha-helix is right-handed). (2) 3. 3 residues per turn (3) Each chain forms hydrogen bonds with the other two
The fibers have diameter between 80 to 160")
Types of collagen Collagen type I i)The fibers have diameter between 80 to 160 nm. ii)Found in bone, dentin, skin, tendon, muscles and walls of blood vessels. Collagen type II i)have a diameter <80 nm ii)found in intervertiberal discs and hyaline cartilage. Collagen type III i)Found in spleen, muscle, and aorta. Collagen type IV Found around different types of in the basement membranes and muscles. Collagen type V It is found in embryonic cell cultures and the basement membranes. Collagen type VI It is found in muscle and skin.

Collagen Amino Acid Composition • Nearly one residue out of three is Gly • Proline content is unusually high • Proline facilitates the formation of the helical conformation of each α-chain because its ring structure causes "kinks" in the peptide chain. • Many modified amino acids are present: – 4 -hydroxyproline – 3 -hydroxyproline – 5 -hydroxylysine The design of collagen. The structural features of collagen ranges from the amino acid sequence, tropocollagen molecules, collagen fibrils to collagen fibers.

BIOSYNTHESIS OF COLLAGEN The polypeptide precursors of the collagen molecule are formed in fibroblasts (or in the related osteoblasts of bone and chondroblasts of cartilage), and are secreted into the extracellular matrix. After enzymatic modification, the mature collagen monomers aggregate and become cross-linked to form collagen fibers.

BIOSYNTHESIS OF COLLAGEN 1. Formation of pro-α chains: Collagen is one of many proteins that normally function outside of cells. Like most proteins produced for export, the newly synthesized polypeptide precursors of α chains (prepro-α chains) contain a special amino acid sequence at their N-terminal ends. This sequence acts as a signal that targets the polypeptide being synthesized for secretion from the cell. The signal sequence facilitates the binding of ribosomes to the rough endoplasmic reticulum (RER), and directs the passage of the prepro-α chain into the lumen of the RER. The signal sequence is rapidly cleaved in the RER to yield a precursor of collagen called a pro-α chain.

2. Hydroxylation: The pro-α chains are processed by a number of enzymic steps within the lumen of the RER while the polypeptides are still being synthesized. Proline and lysine residues found in the Y-position of the –Gly–X–Y– sequence can be hydroxylated to form hydroxyproline and hydroxylysine residues. These hydroxylation reactions require molecular oxygen, Fe 2+, and the reducing agent vitamin C (ascorbic acid), without which the hydroxylating enzymes, prolyl hydroxylase and lysyl hydroxylase, are unable to function. In the case of ascorbic acid deficiency (and, therefore, a lack of prolyl and lysyl hydroxylation), interchain H-bond formation is impaired, as is formation of a stable triple helix. Additionally, collagen fibrils cannot be cross-linked, greatly decreasing the tensile strength of the assembled fiber. The resulting deficiency disease is known as SCURVY. Patients with ascorbic acid deficiency also often show bruises on the limbs as a result of subcutaneous

3. Glycosylation: Some hydroxylysine residues are modified by glycosylation with glucose or glucosylgalactose. 4. Assembly and secretion: After hydroxylation and glycosylation, pro-α chains form procollagen, The formation of procollagen begins with formation of interchain disulfide bonds between the C-terminal extensions of the pro-α chains. This brings the three α chains into an alignment favorable for helix formation. The procollagen molecules move through the Golgi apparatus, where they are packaged in secretory vesicles. The vesicles fuse with the cell membrane, causing the release of procollagen molecules into the extracellular space.

5. Extracellular cleavage of procollagen molecules: After their release, the procollagen molecules are cleaved by N- and Cprocollagen peptidases, which remove the terminal propeptides, releasing triple-helical tropocollagen molecules.

6. Formation of collagen fibrils: Individual tropocollagen molecules spontaneously associate to form collagen fibrils. They form an ordered, overlapping, parallel array, with adjacent collagen molecules arranged in a staggered pattern, each overlapping its neighbor by a length approximately three-quarters of a molecule. 7. Cross-link formation: The fibrillar array of collagen molecules serves as a substrate for lysyl oxidase. This Cu 2+containing extracellular enzyme oxidatively deaminates some of the lysyl and hydroxylysyl residues in collagen. The reactive aldehydes that result (allysine and hydroxyallysine) can condense with lysyl or hydroxy lysyl residues in neighboring collagen molecules to form covalent cross-links and, thus, mature collagen fibers

Inherited Disorders of Connective Tissue Some connective tissue diseases -- often called heritable disorders of connective tissue (HDCTs) -- are the result of changes in certain genes. Many of these are quite rare. Following are some of the more common ones. Ehlers-Danlos syndrome (EDS). Actually a group of more than 10 disorders, EDS is characterized by over-flexible joints, stretchy skin, and abnormal growth of scar tissue. Symptoms can range from mild to disabling. Depending on the specific form of EDS, other symptoms may include: § A curved spine § Weak blood vessels § Bleeding gums § Problems with the lungs, heart valves, or digestion.
. People with EB have skin that is so fragile that it")
Epidermolysis bullosa (EB). People with EB have skin that is so fragile that it tears or blisters as a result of a minor bump, stumble, or even friction from clothing. Some forms of EB may involve the digestive tract, the respiratory tract, the muscles, or the bladder. Caused by defects of several proteins in the skin, EB is usually evident at birth Marfan syndrome affects the bones, ligaments, eyes, heart, and blood vessels. People with Marfan syndrome tend to be tall and have extremely long bones and thin "spider-like" fingers and toes. Other problems may include eye problems due to abnormal placement of the eye lens and enlargement of the aorta (the largest artery in the body), which can lead to a fatal rupture. Marfan syndrome is caused by mutations in the gene that regulates the structure of a protein called fibrillin-1.
- Slides: 14